

Homozygous stop mutation in AHR causes autosomal recessive foveal hypoplasia and infantile nystagmus
- PMID: 31009037
- PMCID: PMC6766433
- DOI: 10.1093/brain/awz098
Homozygous stop mutation in AHR causes autosomal recessive foveal hypoplasia and infantile nystagmus
Abstract
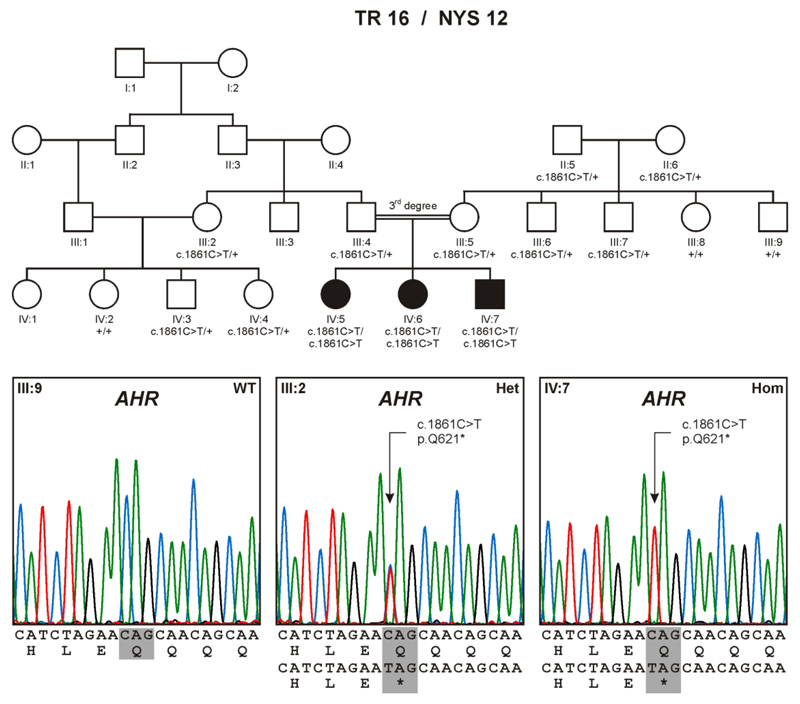
Herein we present a consanguineous family with three children affected by foveal hypoplasia with infantile nystagmus, following an autosomal recessive mode of inheritance. The patients showed normal electroretinography responses, no signs of albinism, and no anterior segment or brain abnormalities. Upon whole exome sequencing, we identified a homozygous mutation (c.1861C>T;p.Q621*) in the aryl hydrocarbon receptor (AHR) gene that perfectly co-segregated with the disease in the larger family. AHR is a ligand-activated transcription factor that has been intensively studied in xenobiotic-induced toxicity. Further, it has been shown to play a physiological role under normal cellular conditions, such as in immunity, inflammatory response and neurogenesis. Notably, knockout of the Ahr gene in mouse impairs optic nerve myelin sheath formation and results in oculomotor deficits sharing many features with our patients: the eye movement disorder in Ahr-/- mice appears early in development and presents as conjugate horizontal pendular nystagmus. We therefore propose AHR to be a novel disease gene for a new, recessively inherited disorder in humans, characterized by infantile nystagmus and foveal hypoplasia.
Keywords: AHR; consanguinity; foveal hypoplasia; nystagmus.
© The Author(s) (2019). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures

Similar articles
-
Crossed VEP asymmetry in a patient with AHR-linked infantile nystagmus and foveal hypoplasia.Doc Ophthalmol. 2024 Aug;149(1):47-52. doi: 10.1007/s10633-024-09979-6. Epub 2024 Jun 26. Doc Ophthalmol. 2024. PMID: 38922562
-
Gene and Protein Expression in Subjects With a Nystagmus-Associated AHR Mutation.Front Genet. 2020 Sep 24;11:582796. doi: 10.3389/fgene.2020.582796. eCollection 2020. Front Genet. 2020. PMID: 33193710 Free PMC article.
-
Novel biallelic AHR splice site mutation cause isolated foveal hypoplasia in Saudi patient: a case report.Ophthalmic Genet. 2022 Jun;43(3):425-429. doi: 10.1080/13816810.2022.2039718. Epub 2022 Feb 21. Ophthalmic Genet. 2022. PMID: 35188035
-
Autosomal dominant foveal hypoplasia without visible macular abnormalities and PAX6 mutations.Jpn J Ophthalmol. 2020 Nov;64(6):635-641. doi: 10.1007/s10384-020-00766-9. Epub 2020 Aug 28. Jpn J Ophthalmol. 2020. PMID: 32857266 Review.
-
Infantile and acquired nystagmus in childhood.Eur J Paediatr Neurol. 2012 Nov;16(6):567-72. doi: 10.1016/j.ejpn.2012.02.010. Epub 2012 Mar 28. Eur J Paediatr Neurol. 2012. PMID: 22459007 Review.
Cited by
-
Unraveling the genetic cause of hereditary ophthalmic disorders in Arab societies from Israel and the Palestinian Authority.Eur J Hum Genet. 2020 Jun;28(6):742-753. doi: 10.1038/s41431-019-0566-3. Epub 2020 Jan 2. Eur J Hum Genet. 2020. PMID: 31896775 Free PMC article.
-
SLC38A8 mutations result in arrested retinal development with loss of cone photoreceptor specialization.Hum Mol Genet. 2020 Nov 4;29(18):2989-3002. doi: 10.1093/hmg/ddaa166. Hum Mol Genet. 2020. PMID: 32744312 Free PMC article.
-
Proper modulation of AHR signaling is necessary for establishing neural connectivity and oligodendrocyte precursor cell development in the embryonic zebrafish brain.Front Mol Neurosci. 2022 Nov 29;15:1032302. doi: 10.3389/fnmol.2022.1032302. eCollection 2022. Front Mol Neurosci. 2022. PMID: 36523606 Free PMC article.
-
Diet-Host-Microbiota Interactions Shape Aryl Hydrocarbon Receptor Ligand Production to Modulate Intestinal Homeostasis.Annu Rev Nutr. 2021 Oct 11;41:455-478. doi: 10.1146/annurev-nutr-043020-090050. Annu Rev Nutr. 2021. PMID: 34633858 Free PMC article. Review.
-
Comparison of OCT imaging in children with foveal hypoplasia born full term versus preterm.Graefes Arch Clin Exp Ophthalmol. 2022 Sep;260(9):3075-3085. doi: 10.1007/s00417-022-05664-z. Epub 2022 Apr 21. Graefes Arch Clin Exp Ophthalmol. 2022. PMID: 35445879
References
-
- Azuma N, Nishina S, Yanagisawa H, Okuyama T, Yamada M. PAX6 missense mutation in isolated foveal hypoplasia. Nat Genet. 1996;13:141–2. - PubMed
-
- Bassi MT, Schiaffino MV, Renieri A, De Nigris F, Galli L, Bruttini M, Gebbia M, Bergen AA, Lewis RA, Ballabio A. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat Genet. 1995;10:13–9. - PubMed
-
- Choudhuri I, Sarvananthan N, Gottlob I. Survey of management of acquired nystagmus in the United Kingdom. Eye (Lond) 2007;21:1194–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
