

Expression estimation and eQTL mapping for HLA genes with a personalized pipeline
- PMID: 31009447
- PMCID: PMC6497317
- DOI: 10.1371/journal.pgen.1008091
Expression estimation and eQTL mapping for HLA genes with a personalized pipeline
Abstract
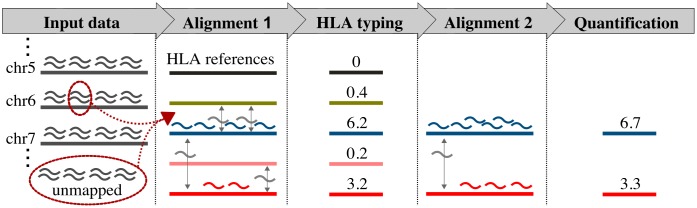
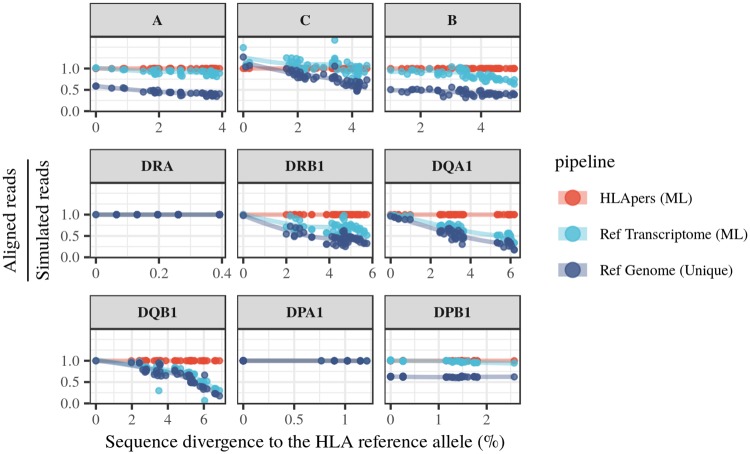
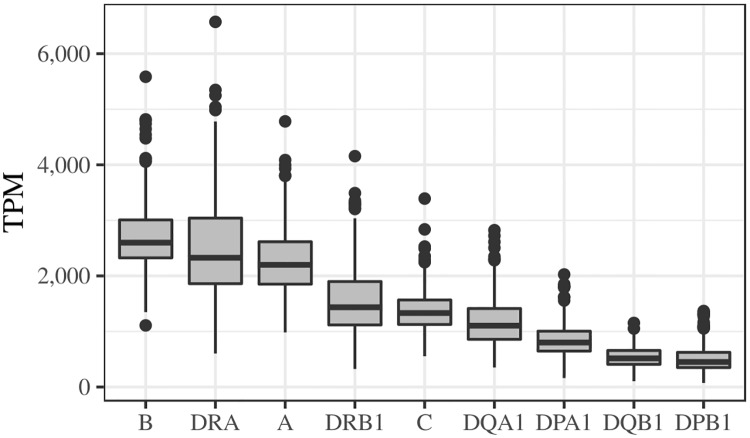
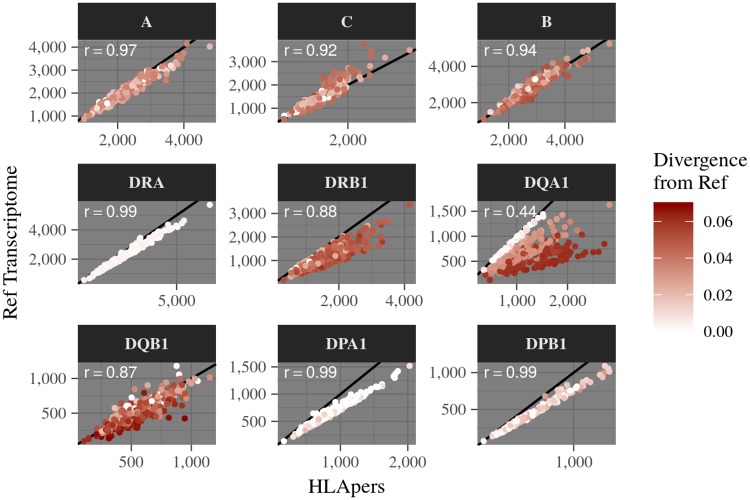
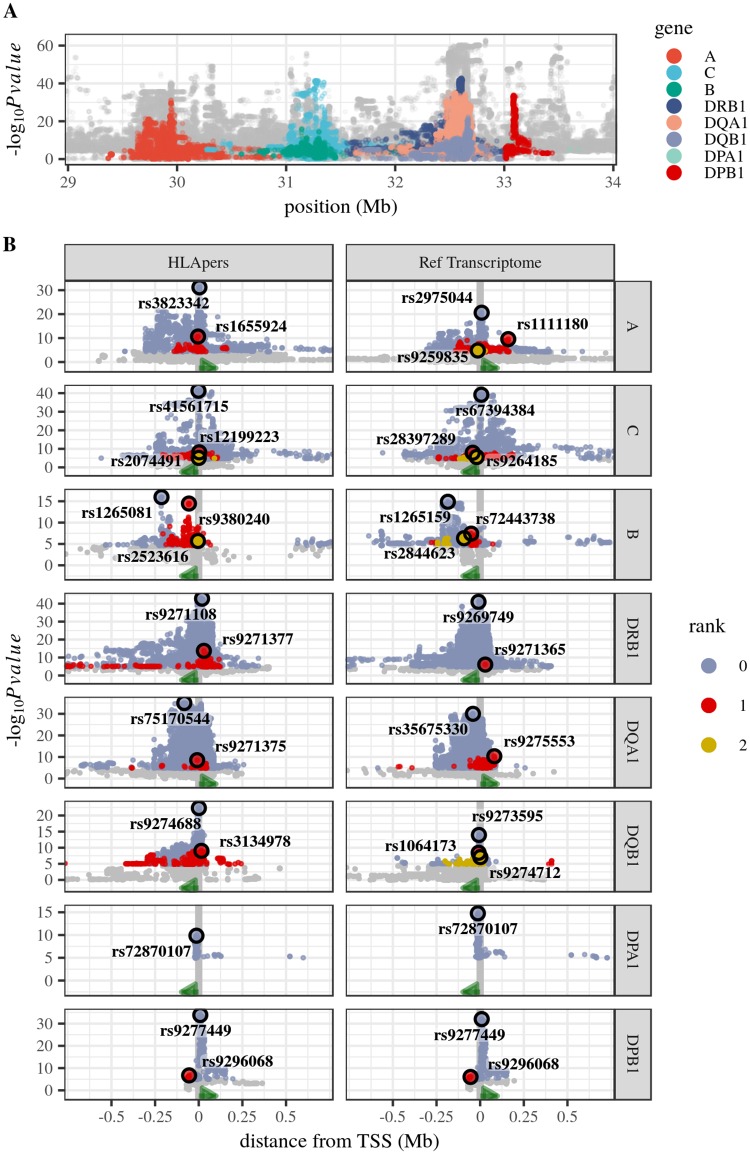
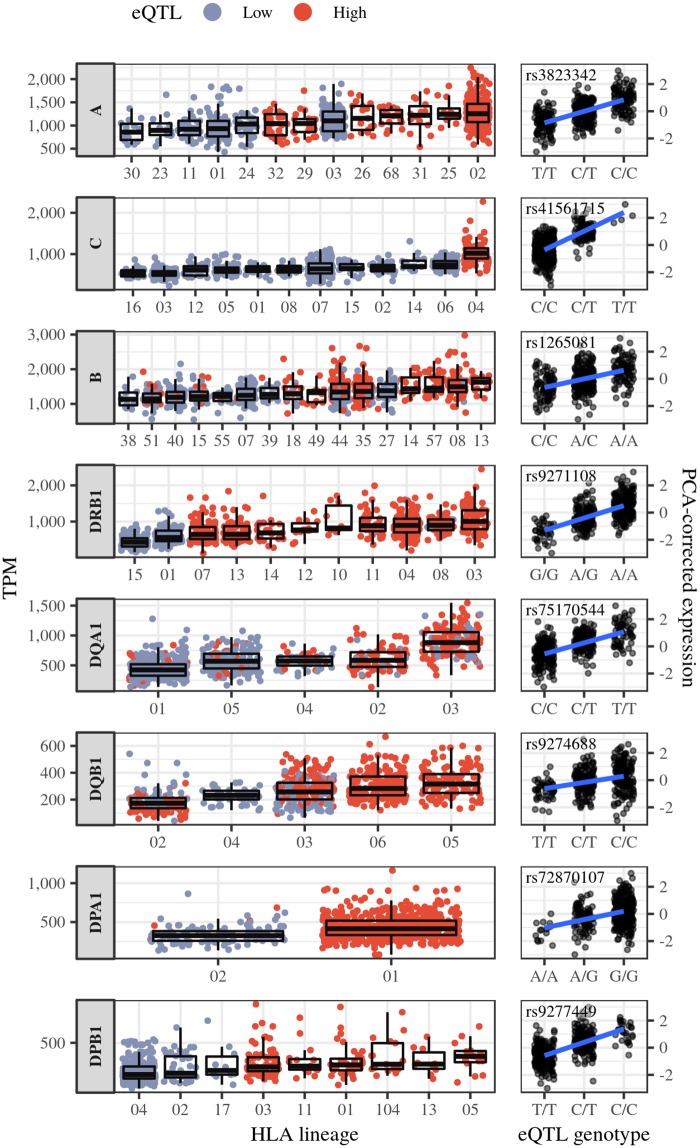
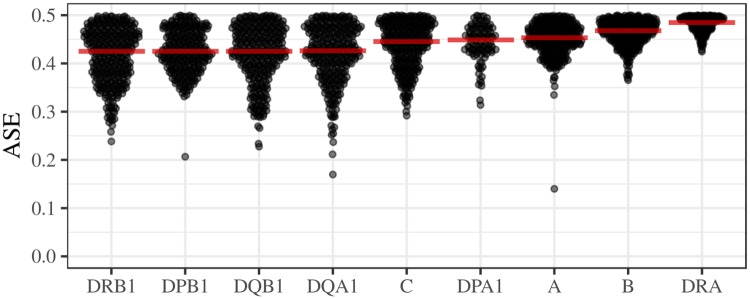
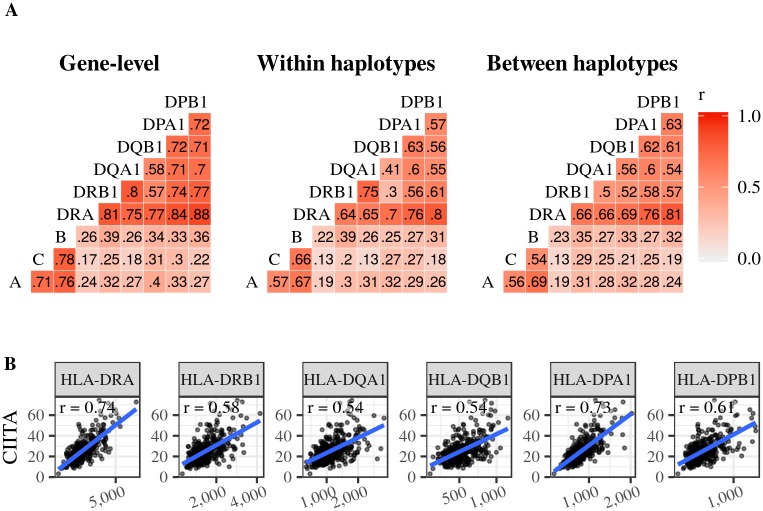
The HLA (Human Leukocyte Antigens) genes are well-documented targets of balancing selection, and variation at these loci is associated with many disease phenotypes. Variation in expression levels also influences disease susceptibility and resistance, but little information exists about the regulation and population-level patterns of expression. This results from the difficulty in mapping short reads originated from these highly polymorphic loci, and in accounting for the existence of several paralogues. We developed a computational pipeline to accurately estimate expression for HLA genes based on RNA-seq, improving both locus-level and allele-level estimates. First, reads are aligned to all known HLA sequences in order to infer HLA genotypes, then quantification of expression is carried out using a personalized index. We use simulations to show that expression estimates obtained in this way are not biased due to divergence from the reference genome. We applied our pipeline to the GEUVADIS dataset, and compared the quantifications to those obtained with reference transcriptome. Although the personalized pipeline recovers more reads, we found that using the reference transcriptome produces estimates similar to the personalized pipeline (r ≥ 0.87) with the exception of HLA-DQA1. We describe the impact of the HLA-personalized approach on downstream analyses for nine classical HLA loci (HLA-A, HLA-C, HLA-B, HLA-DRA, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, HLA-DPB1). Although the influence of the HLA-personalized approach is modest for eQTL mapping, the p-values and the causality of the eQTLs obtained are better than when the reference transcriptome is used. We investigate how the eQTLs we identified explain variation in expression among lineages of HLA alleles. Finally, we discuss possible causes underlying differences between expression estimates obtained using RNA-seq, antibody-based approaches and qPCR.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Meyer D, Single R, Mack S, Lancaster A, Nelson M, Erlich H, et al. Single locus polymorphism of classical HLA genes. In: Hansen JA Immunobiology of the Human MHC: Proceedings of the 13th International Histocompatibility Workshop and Conference. Seattle, WA: IHWG Press; 2007. p. 653–704.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
