

Molecular mechanisms in progression of HPV-associated cervical carcinogenesis
- PMID: 31014351
- PMCID: PMC6477741
- DOI: 10.1186/s12929-019-0520-2
Molecular mechanisms in progression of HPV-associated cervical carcinogenesis
Retraction in
-
Retraction Note: Molecular mechanisms in progression of HPV-associated cervical carcinogenesis.J Biomed Sci. 2019 Jul 4;26(1):50. doi: 10.1186/s12929-019-0545-6. J Biomed Sci. 2019. PMID: 31272503 Free PMC article.
Abstract
Cervical cancer is the fourth most frequent cancer in women worldwide and a major cause of mortality in developing countries. Persistent infection with high-risk human papillomavirus (HPV) is a necessary cause for the development of cervical cancer. In addition, genetic and epigenetic alterations in host cell genes are crucial for progression of cervical precancerous lesions to invasive cancer. Although much progress has been made in understanding the life cycle of HPV and it's role in the development of cervical cancer, there is still a critical need for accurate surveillance strategies and targeted therapeutic options to eradicate these cancers in patients. Given the widespread nature of HPV infection and the type specificity of currently available HPV vaccines, it is crucial that molecular details of the natural history of HPV infection as well as the biological activities of viral oncoproteins be elucidated. A better understanding of the mechanisms involved in oncogenesis can provide novel insights and opportunities for designing effective therapeutic approaches against HPV-associated malignancies. In this review, we briefly summarize epigenetic alterations and events that cause alterations in host genomes inducing cell cycle deregulation, aberrant proliferation and genomic instability contributing to tumorigenesis.
Keywords: Cervical carcinogenesis; Chromosomal mutations; Epigenetic alterations; Oncogene expression.
Conflict of interest statement
Authors’ information
None.
Ethics approval and consent to participate
Not applicable (The present paper does not report on or involve the use of any animal or human data or tissue).
Consent for publication
Not applicable (The present paper does not contain data from any individual person).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
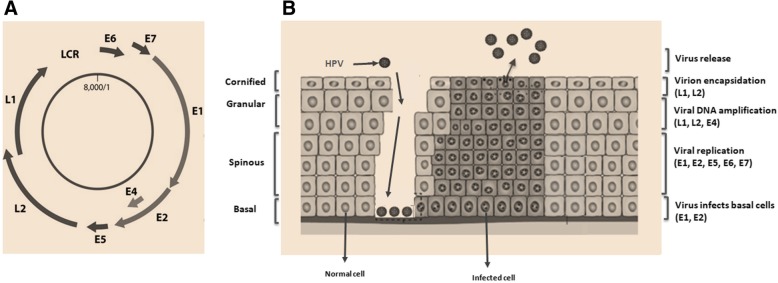
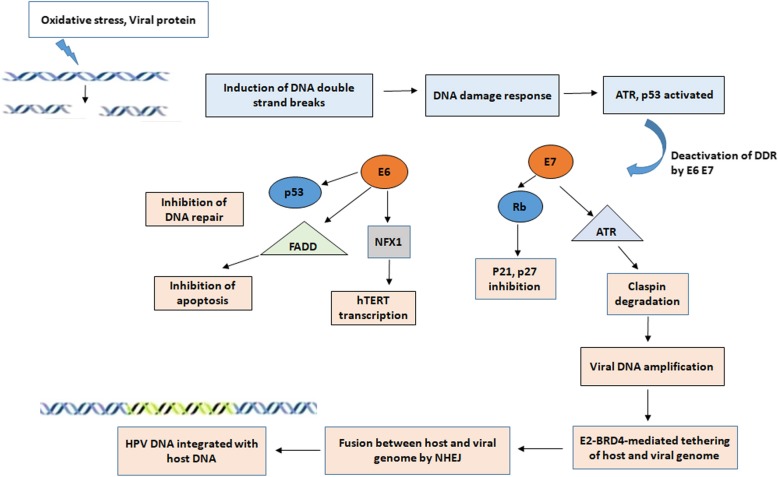
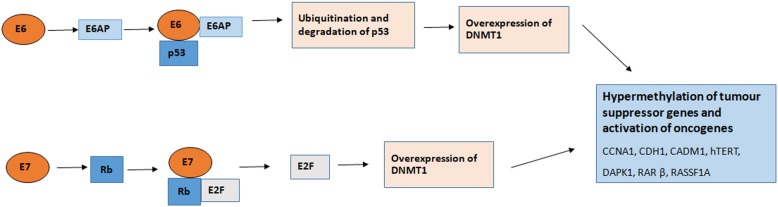
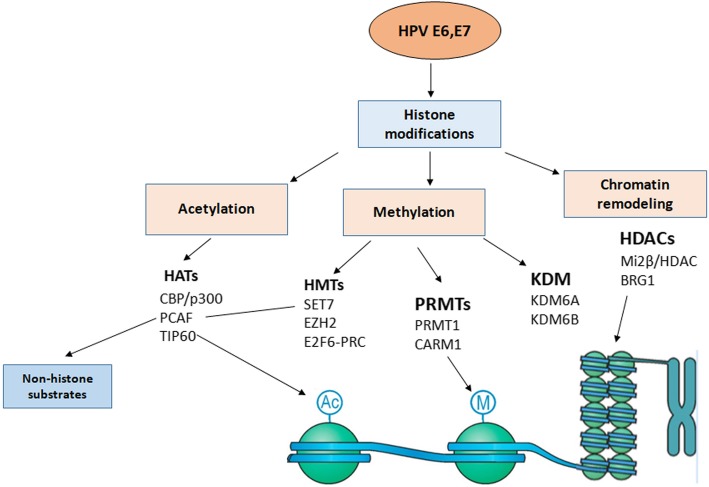
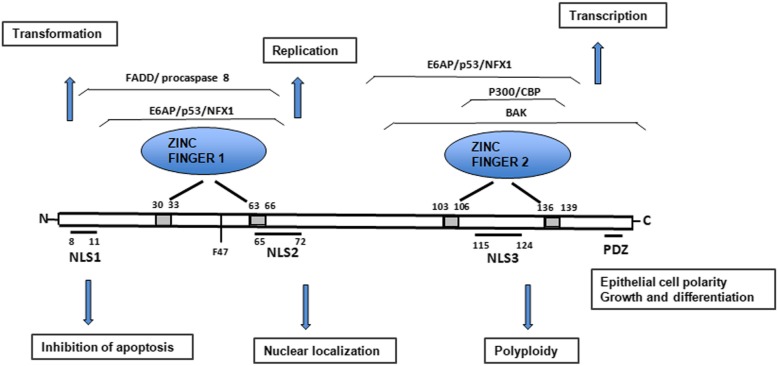
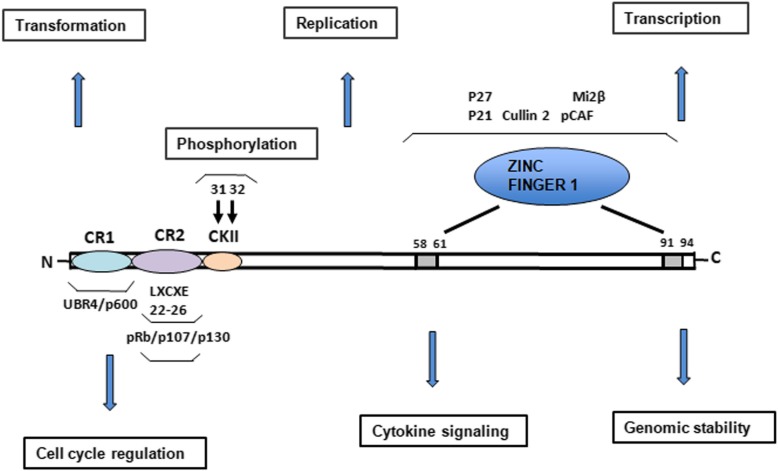
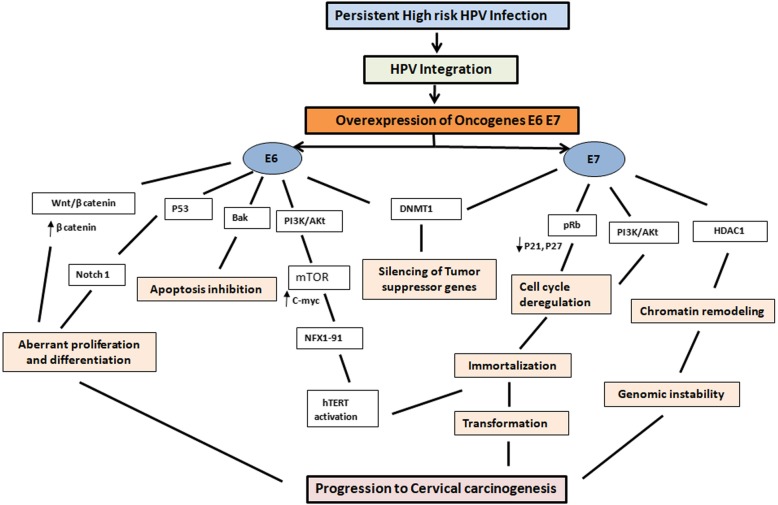
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
