

Modeling Protein Destiny in Developing Fruit
- PMID: 31015299
- PMCID: PMC6752906
- DOI: 10.1104/pp.19.00086
Modeling Protein Destiny in Developing Fruit
Abstract
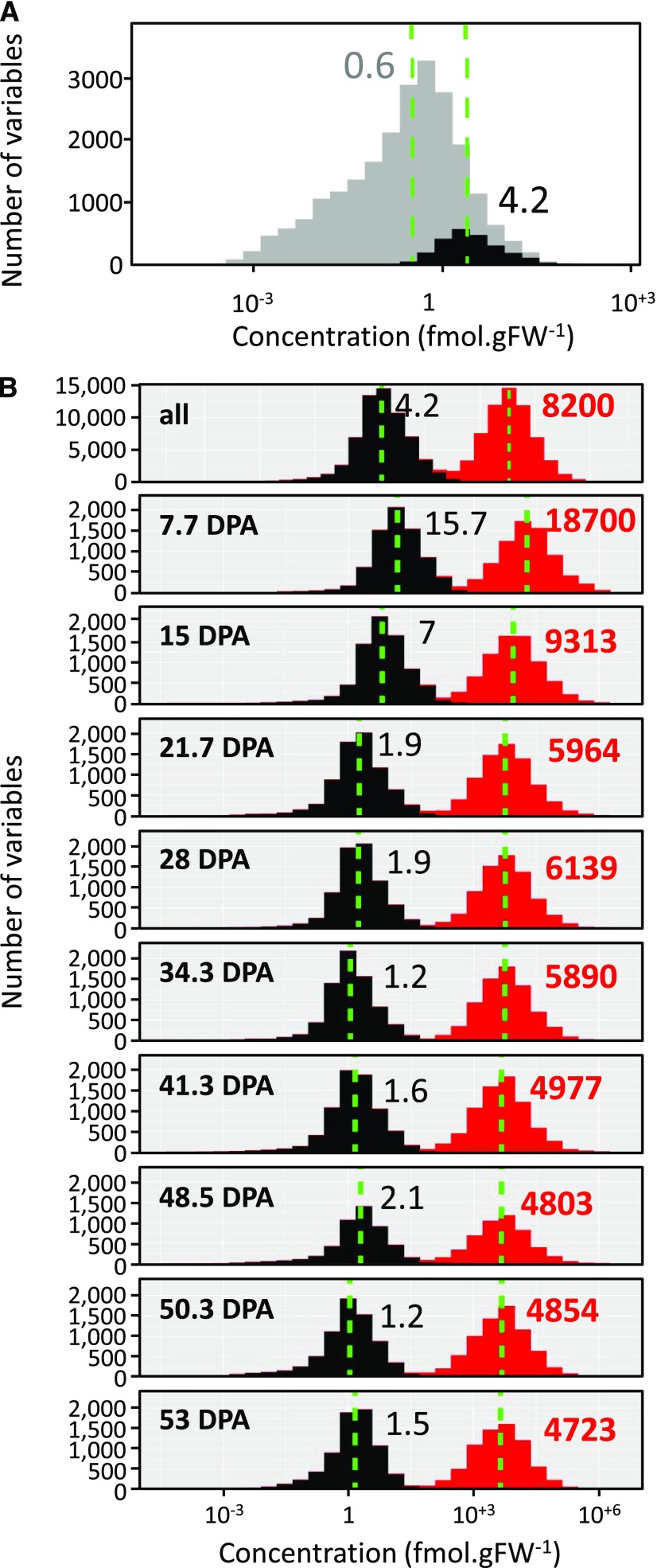
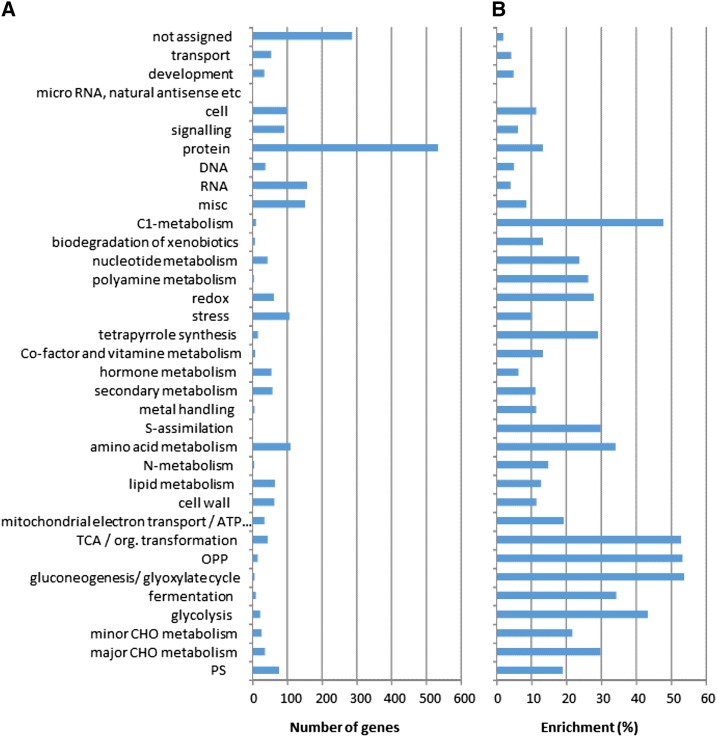
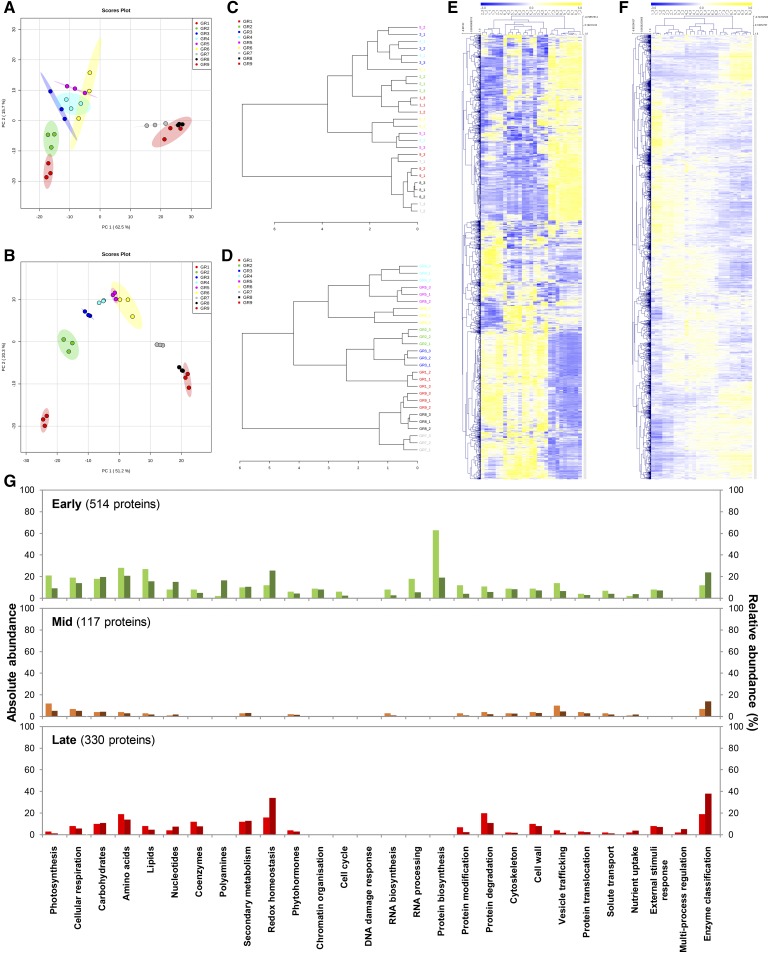
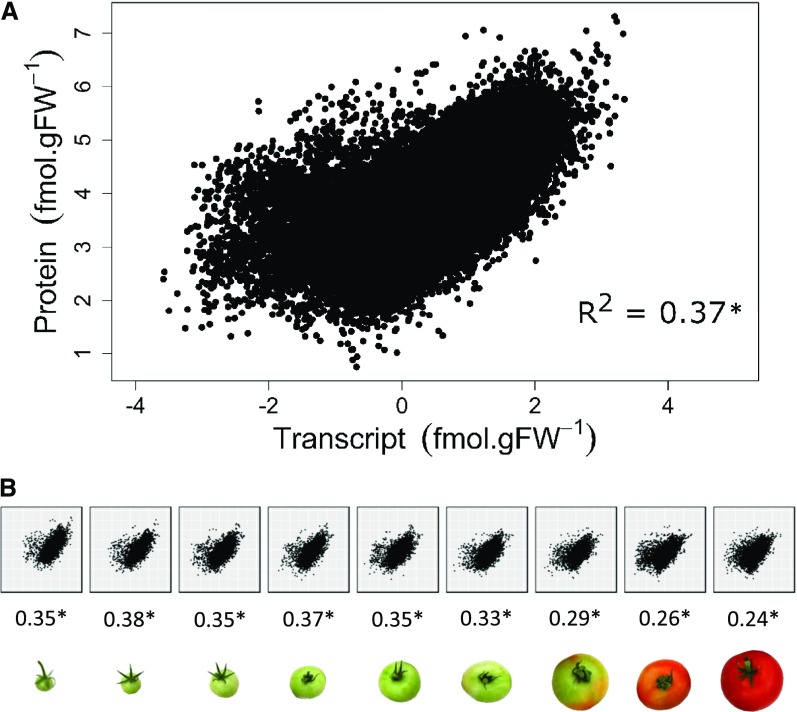
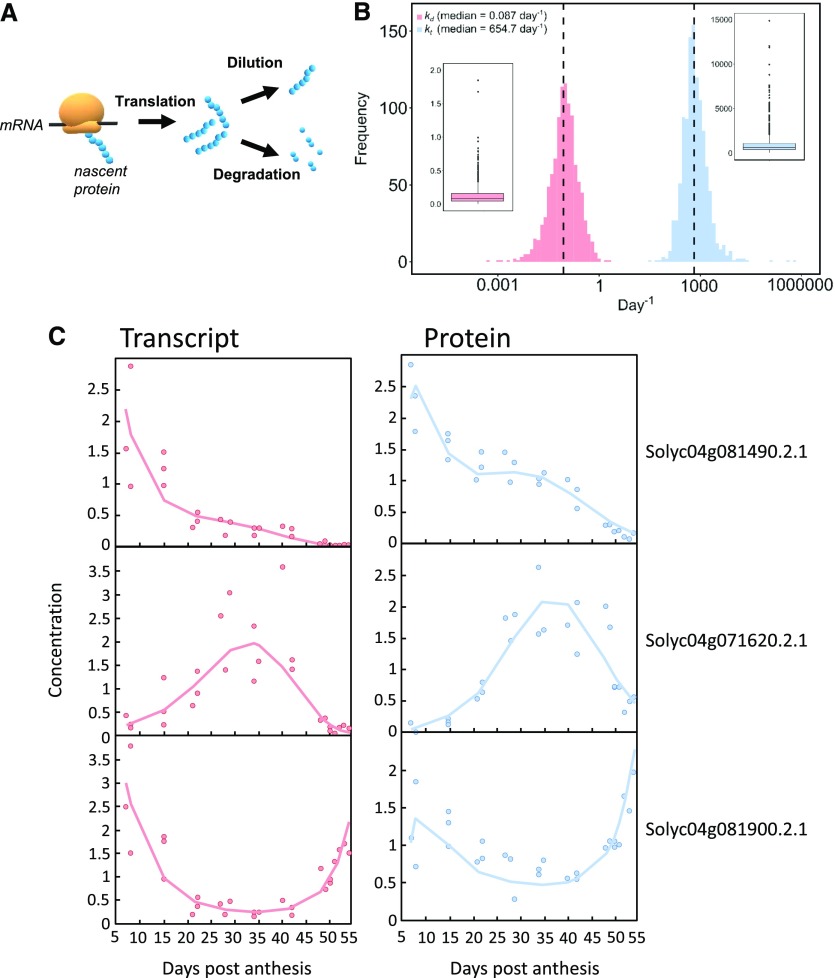
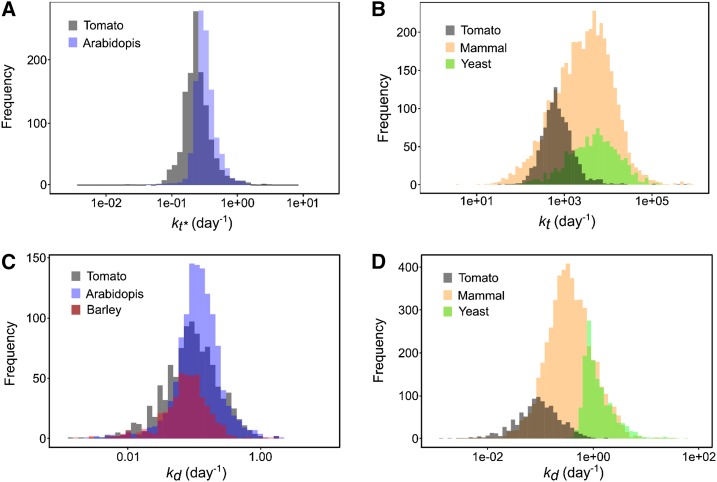
Protein synthesis and degradation are essential processes that regulate cell status. Because labeling in bulky organs, such as fruits, is difficult, we developed a modeling approach to study protein turnover at the global scale in developing tomato (Solanum lycopersicum) fruit. Quantitative data were collected for transcripts and proteins during fruit development. Clustering analysis showed smaller changes in protein abundance compared to mRNA abundance. Furthermore, protein and transcript abundance were poorly correlated, and the coefficient of correlation decreased during fruit development and ripening, with transcript levels decreasing more than protein levels. A mathematical model with one ordinary differential equation was used to estimate translation (kt ) and degradation (kd ) rate constants for almost 2,400 detected transcript-protein pairs and was satisfactorily fitted for >1,000 pairs. The model predicted median values of ∼2 min for the translation of a protein, and a protein lifetime of ∼11 d. The constants were validated and inspected for biological relevance. Proteins involved in protein synthesis had higher kt and kd values, indicating that the protein machinery is particularly flexible. Our model also predicts that protein concentration is more strongly affected by the rate of translation than that of degradation.
© 2019 American Society of Plant Biologists. All Rights Reserved.
Figures
References
-
- Andrews S. (2010). FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (January 1, 2010)
-
- Beauvoit BP, Colombié S, Monier A, Andrieu M-H, Biais B, Bénard C, Chéniclet C, Dieuaide-Noubhani M, Nazaret C, Mazat J-P, et al. (2014) Model-assisted analysis of sugar metabolism throughout tomato fruit development reveals enzyme and carrier properties in relation to vacuole expansion. Plant Cell 26: 3224–3242 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
