

A DNMT3A PWWP mutation leads to methylation of bivalent chromatin and growth retardation in mice
- PMID: 31015495
- PMCID: PMC6478690
- DOI: 10.1038/s41467-019-09713-w
A DNMT3A PWWP mutation leads to methylation of bivalent chromatin and growth retardation in mice
Abstract
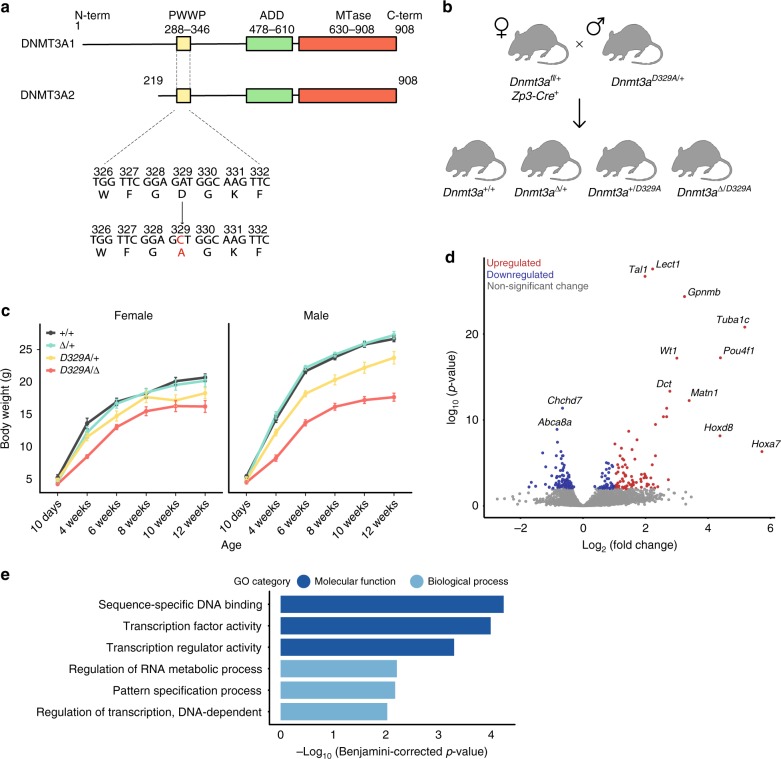
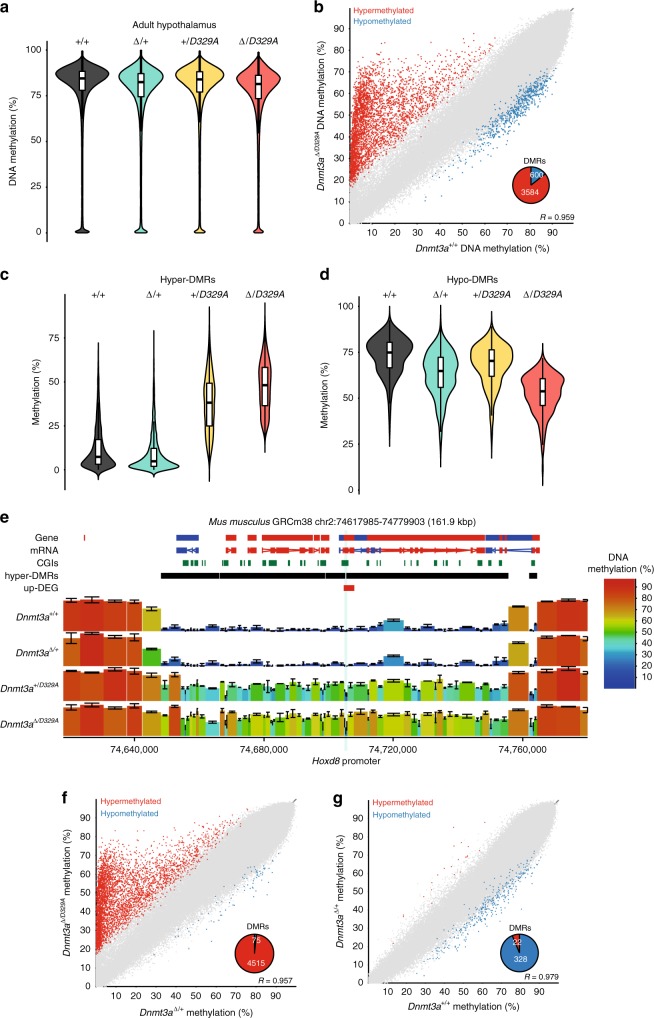
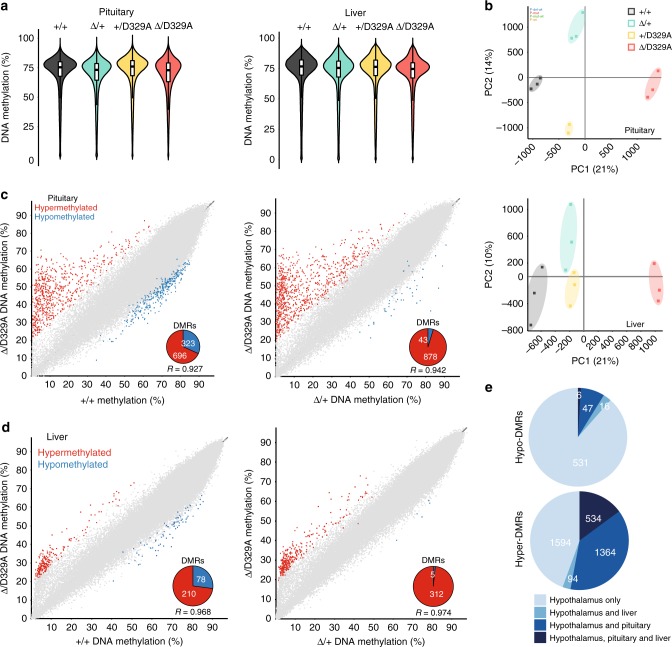
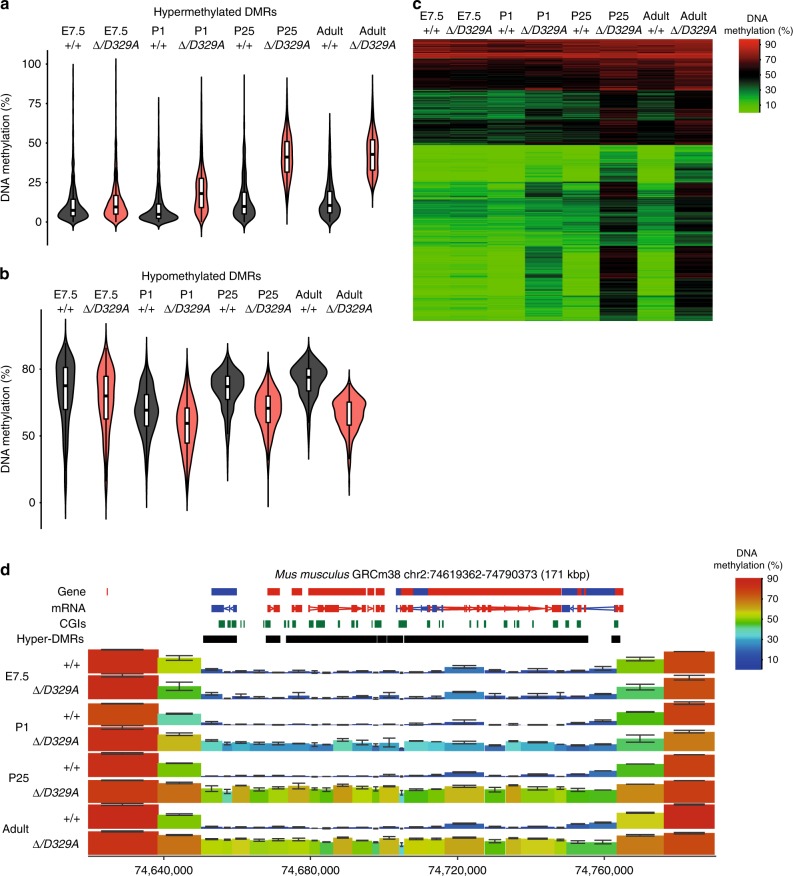
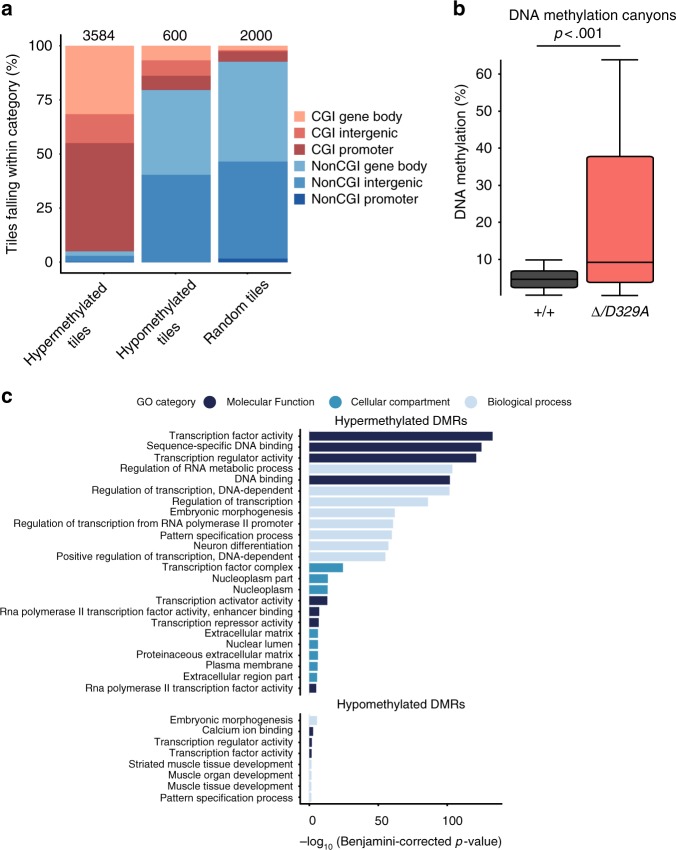
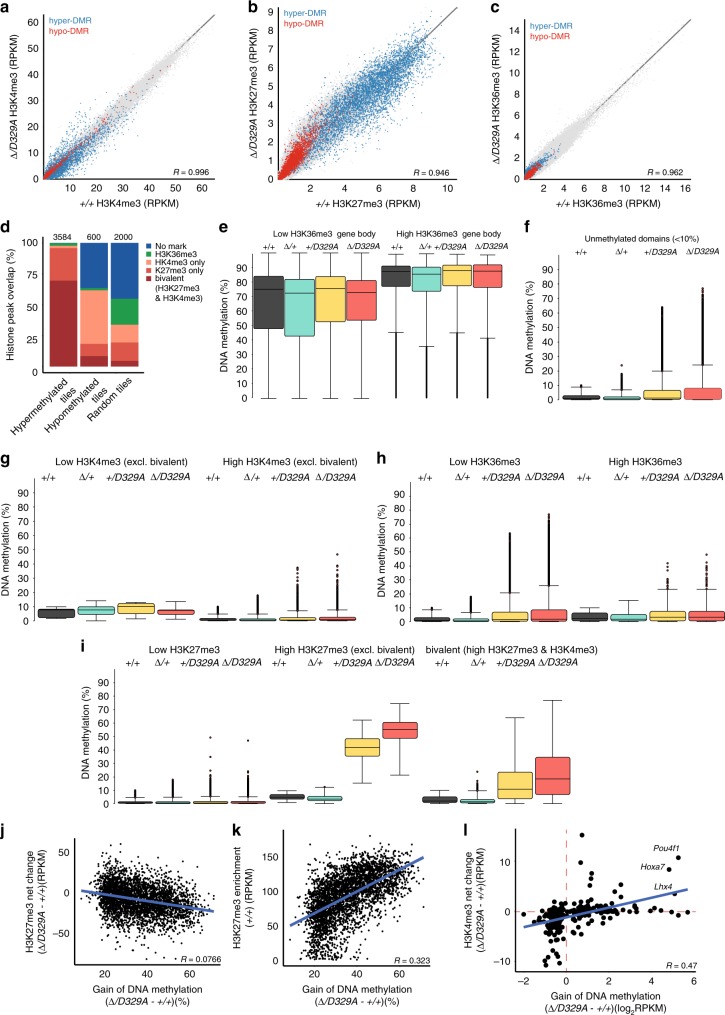
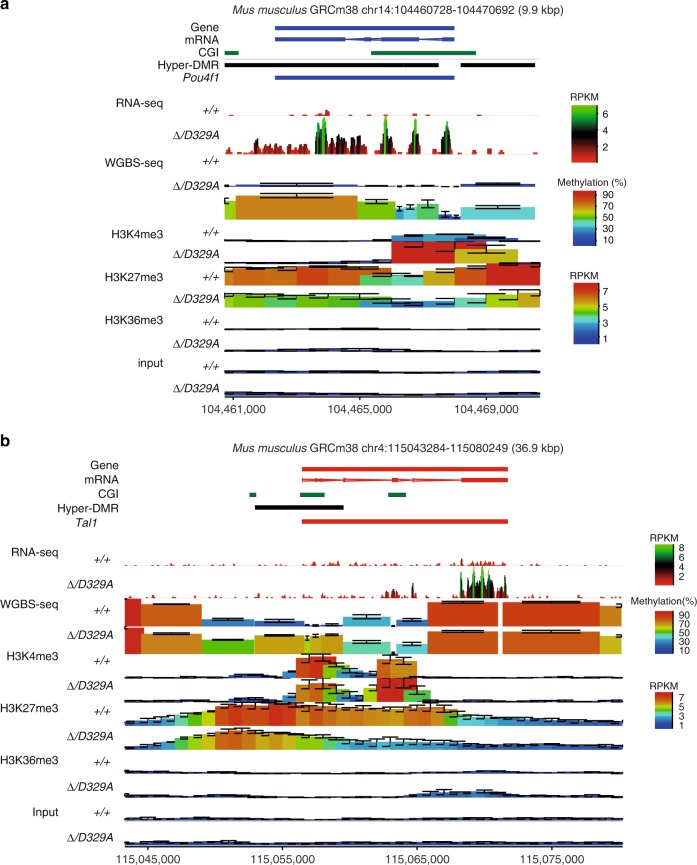
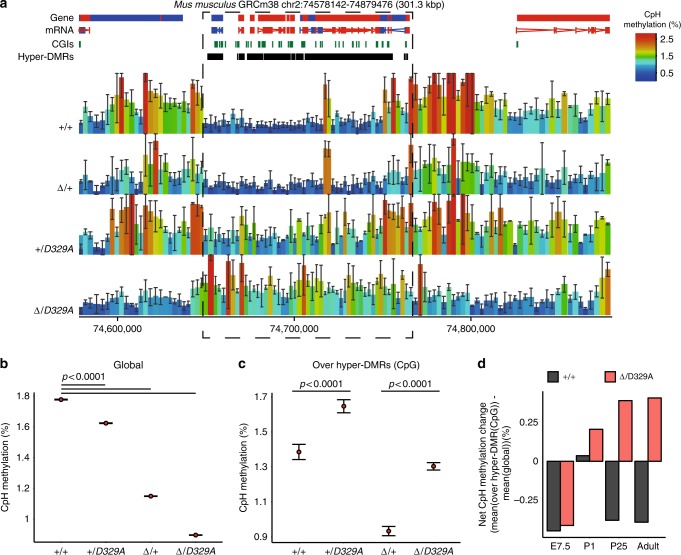
DNA methyltransferases (DNMTs) deposit DNA methylation, which regulates gene expression and is essential for mammalian development. Histone post-translational modifications modulate the recruitment and activity of DNMTs. The PWWP domains of DNMT3A and DNMT3B are posited to interact with histone 3 lysine 36 trimethylation (H3K36me3); however, the functionality of this interaction for DNMT3A remains untested in vivo. Here we present a mouse model carrying a D329A point mutation in the DNMT3A PWWP domain. The mutation causes dominant postnatal growth retardation. At the molecular level, it results in progressive DNA hypermethylation across domains marked by H3K27me3 and bivalent chromatin, and de-repression of developmental regulatory genes in adult hypothalamus. Evaluation of non-CpG methylation, a marker of de novo methylation, further demonstrates the altered recruitment and activity of DNMT3AD329A at bivalent domains. This work provides key molecular insights into the function of the DNMT3A-PWWP domain and role of DNMT3A in regulating postnatal growth.
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
The DNMT3A PWWP domain is essential for the normal DNA methylation landscape in mouse somatic cells and oocytes.PLoS Genet. 2021 May 28;17(5):e1009570. doi: 10.1371/journal.pgen.1009570. eCollection 2021 May. PLoS Genet. 2021. PMID: 34048432 Free PMC article.
-
Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions.Nat Genet. 2019 Jan;51(1):96-105. doi: 10.1038/s41588-018-0274-x. Epub 2018 Nov 26. Nat Genet. 2019. PMID: 30478443 Free PMC article.
-
Both combinatorial K4me0-K36me3 marks on sister histone H3s of a nucleosome are required for Dnmt3a-Dnmt3L mediated de novo DNA methylation.J Genet Genomics. 2020 Feb 20;47(2):105-114. doi: 10.1016/j.jgg.2019.12.006. Epub 2020 Feb 21. J Genet Genomics. 2020. PMID: 32173286
-
Using human disease mutations to understand de novo DNA methyltransferase function.Biochem Soc Trans. 2024 Oct 30;52(5):2059-2075. doi: 10.1042/BST20231017. Biochem Soc Trans. 2024. PMID: 39446312 Free PMC article. Review.
-
Molecular coupling of DNA methylation and histone methylation.Epigenomics. 2010 Oct;2(5):657-69. doi: 10.2217/epi.10.44. Epigenomics. 2010. PMID: 21339843 Free PMC article. Review.
Cited by
-
Interplay between chromatin marks in development and disease.Nat Rev Genet. 2022 Mar;23(3):137-153. doi: 10.1038/s41576-021-00416-x. Epub 2021 Oct 4. Nat Rev Genet. 2022. PMID: 34608297 Review.
-
Novel DNMT3A Germline Variant in a Patient with Multiple Paragangliomas and Papillary Thyroid Carcinoma.Cancers (Basel). 2020 Nov 9;12(11):3304. doi: 10.3390/cancers12113304. Cancers (Basel). 2020. PMID: 33182397 Free PMC article.
-
A High-Fat Diet Induces Epigenetic 1-Carbon Metabolism, Homocystinuria, and Renal-Dependent HFpEF.Nutrients. 2025 Jan 8;17(2):216. doi: 10.3390/nu17020216. Nutrients. 2025. PMID: 39861346 Free PMC article.
-
The DNMT3A PWWP domain is essential for the normal DNA methylation landscape in mouse somatic cells and oocytes.PLoS Genet. 2021 May 28;17(5):e1009570. doi: 10.1371/journal.pgen.1009570. eCollection 2021 May. PLoS Genet. 2021. PMID: 34048432 Free PMC article.
-
Alterations to DNMT3A in Hematologic Malignancies.Cancer Res. 2021 Jan 15;81(2):254-263. doi: 10.1158/0008-5472.CAN-20-3033. Epub 2020 Oct 21. Cancer Res. 2021. PMID: 33087320 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
