

Extensive deep neural networks for transferring small scale learning to large scale systems
- PMID: 31015950
- PMCID: PMC6460955
- DOI: 10.1039/c8sc04578j
Extensive deep neural networks for transferring small scale learning to large scale systems
Abstract
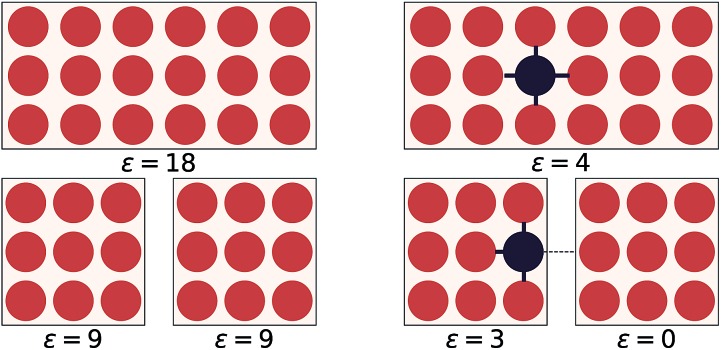
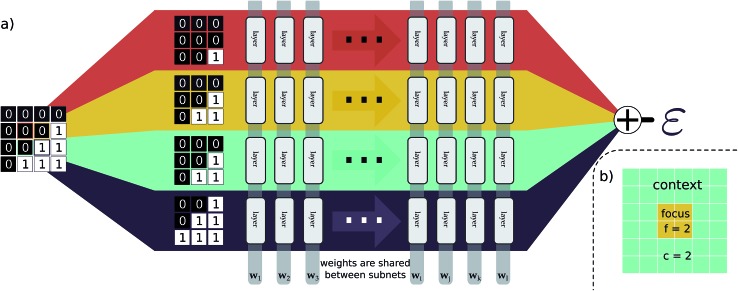
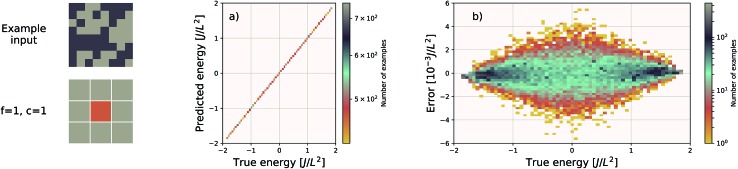
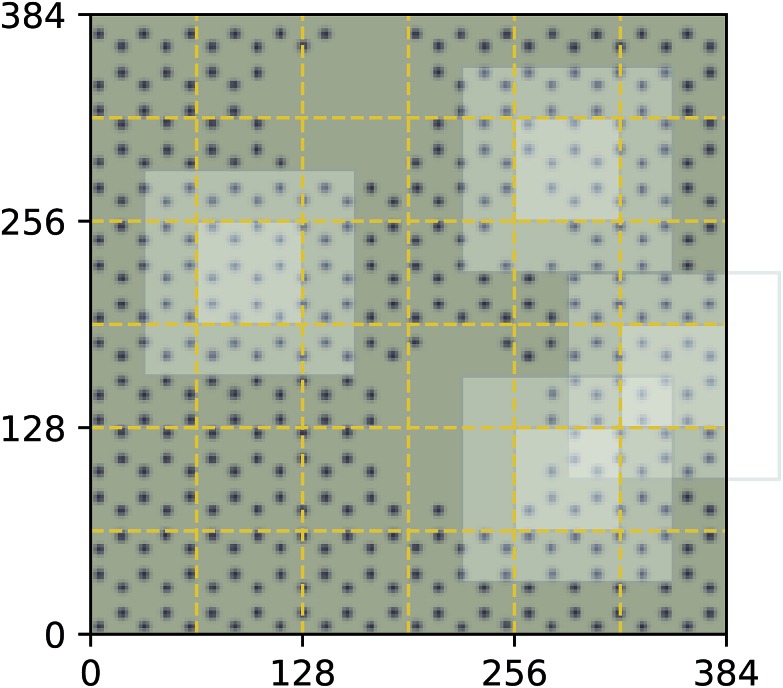
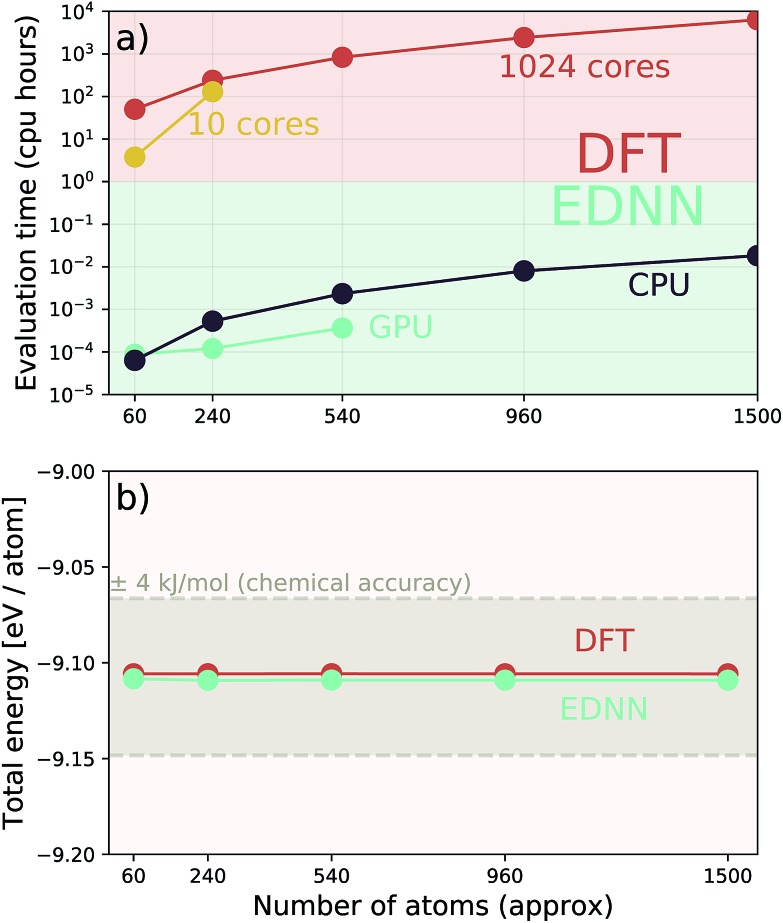
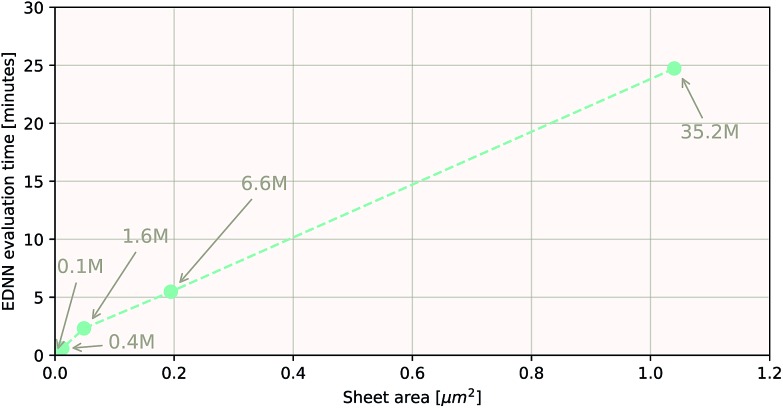
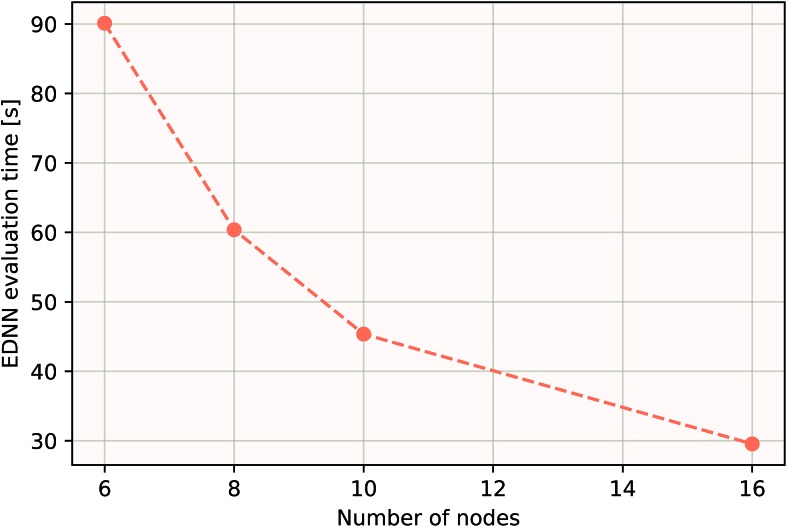
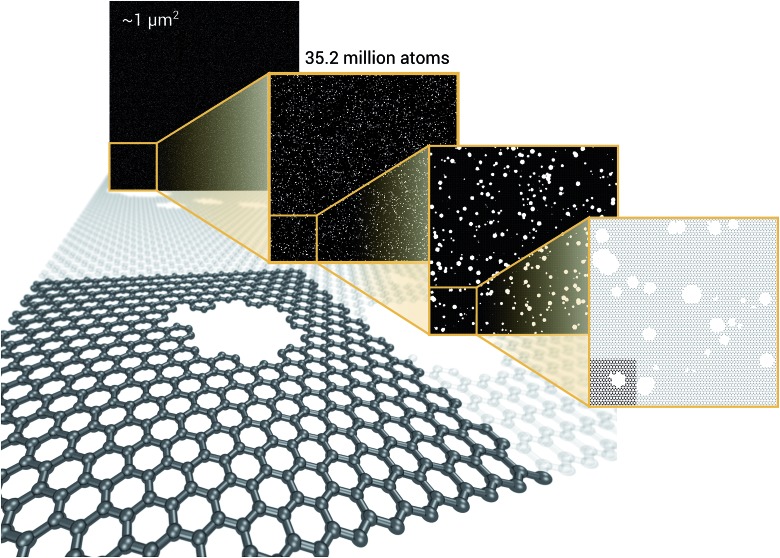
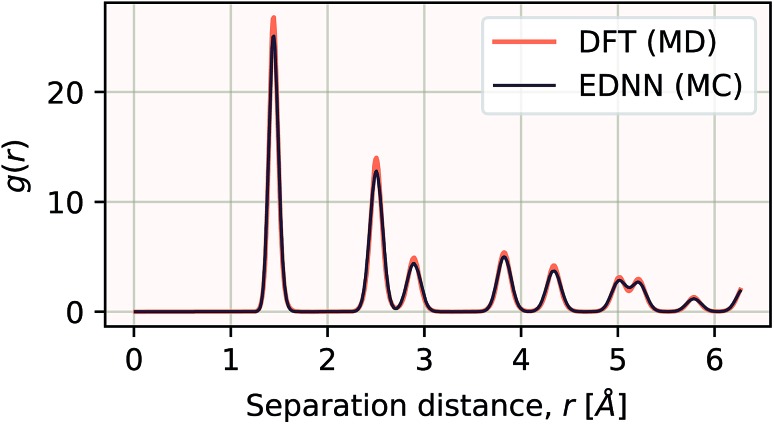
We present a physically-motivated topology of a deep neural network that can efficiently infer extensive parameters (such as energy, entropy, or number of particles) of arbitrarily large systems, doing so with scaling. We use a form of domain decomposition for training and inference, where each sub-domain (tile) is comprised of a non-overlapping focus region surrounded by an overlapping context region. The size of these regions is motivated by the physical interaction length scales of the problem. We demonstrate the application of EDNNs to three physical systems: the Ising model and two hexagonal/graphene-like datasets. In the latter, an EDNN was able to make total energy predictions of a 60 atoms system, with comparable accuracy to density functional theory (DFT), in 57 milliseconds. Additionally EDNNs are well suited for massively parallel evaluation, as no communication is necessary during neural network evaluation. We demonstrate that EDNNs can be used to make an energy prediction of a two-dimensional 35.2 million atom system, over 1.0 μm2 of material, at an accuracy comparable to DFT, in under 25 minutes. Such a system exists on a length scale visible with optical microscopy and larger than some living organisms.
Figures
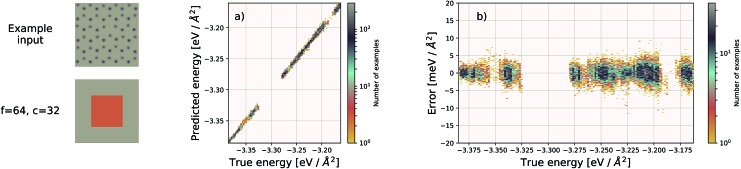
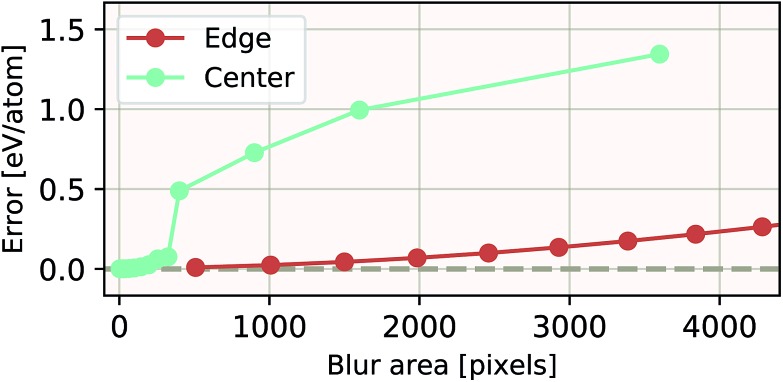
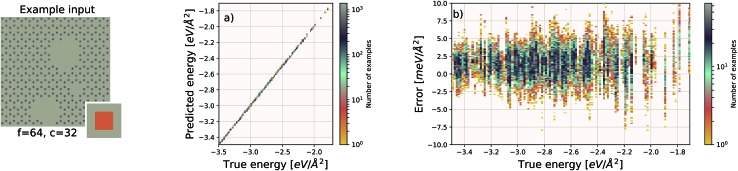
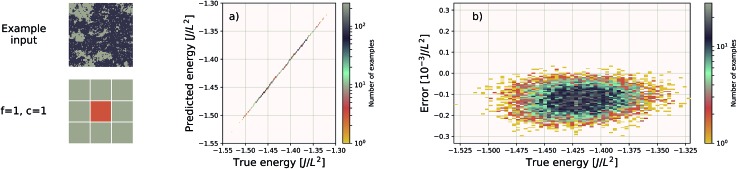
References
-
- Chetlur S. and Woolley C., arXiv, 2014, 1–9.
-
- Lacey G., Taylor G. W. and Areibi S., arXiv, 2016.
-
- Jia Y., Shelhamer E., Donahue J., Karayev S., Long J., Girshick R., Guadarrama S. and Darrell T., arXiv, 2014, 675–678..
-
- Silver D., Huang A., Maddison C. J., Guez A., Sifre L., van den Driessche G., Schrittwieser J., Antonoglou I., Panneershelvam V., Lanctot M., Dieleman S., Grewe D., Nham J., Kalchbrenner N., Sutskever I., Lillicrap T., Leach M., Kavukcuoglu K., Graepel T., Hassabis D. Nature. 2016;529:484–489. - PubMed
-
- Mnih V., Kavukcuoglu K., Silver D., Rusu A. a., Veness J., Bellemare M. G., Graves A., Riedmiller M., Fidjeland A. K., Ostrovski G., Petersen S., Beattie C., Sadik A., Antonoglou I., King H., Kumaran D., Wierstra D., Legg S., Hassabis D. Nature. 2015;518:529–533. - PubMed
LinkOut - more resources
Full Text Sources