

Bioinformatic and mutational studies of related toxin-antitoxin pairs in Mycobacterium tuberculosis predict and identify key functional residues
- PMID: 31018964
- PMCID: PMC6556569
- DOI: 10.1074/jbc.RA118.006814
Bioinformatic and mutational studies of related toxin-antitoxin pairs in Mycobacterium tuberculosis predict and identify key functional residues
Abstract
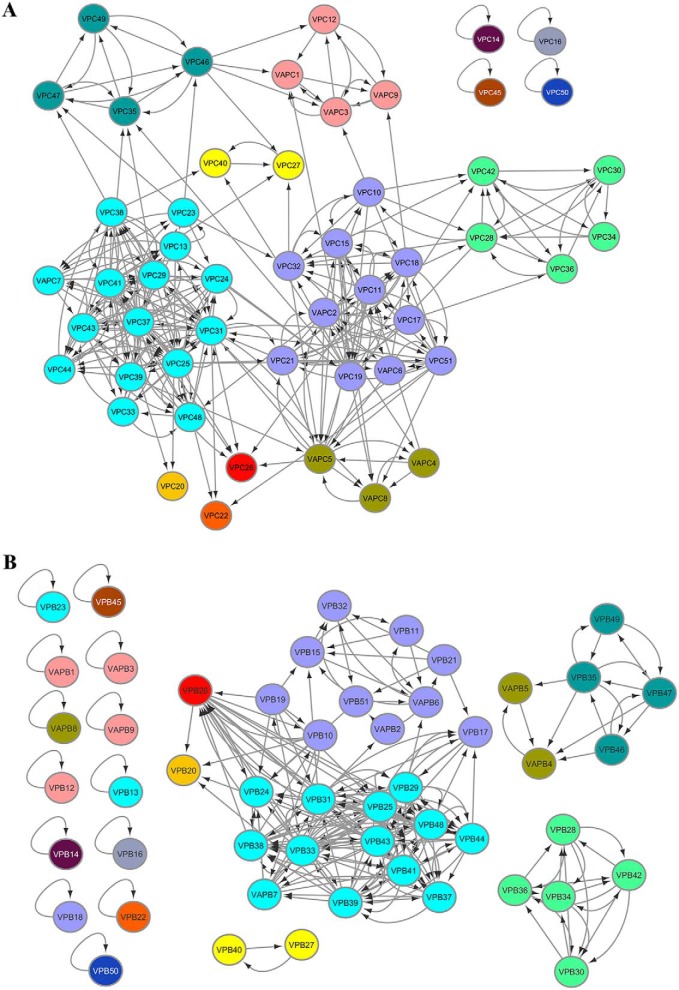
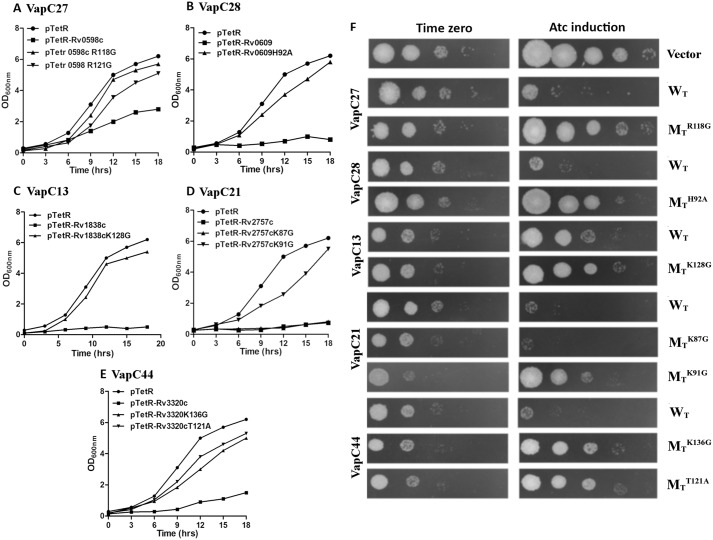
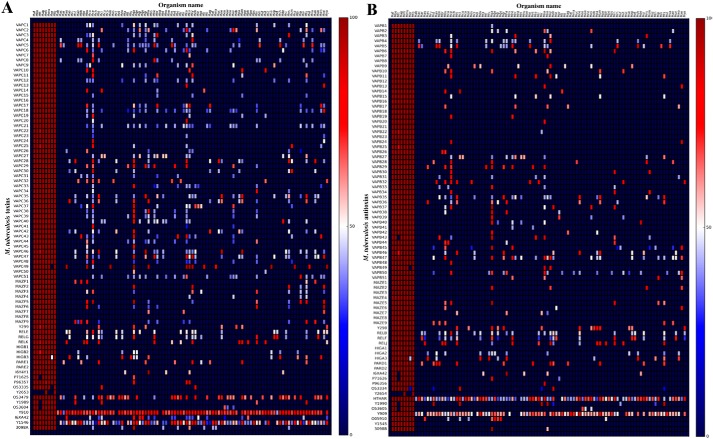
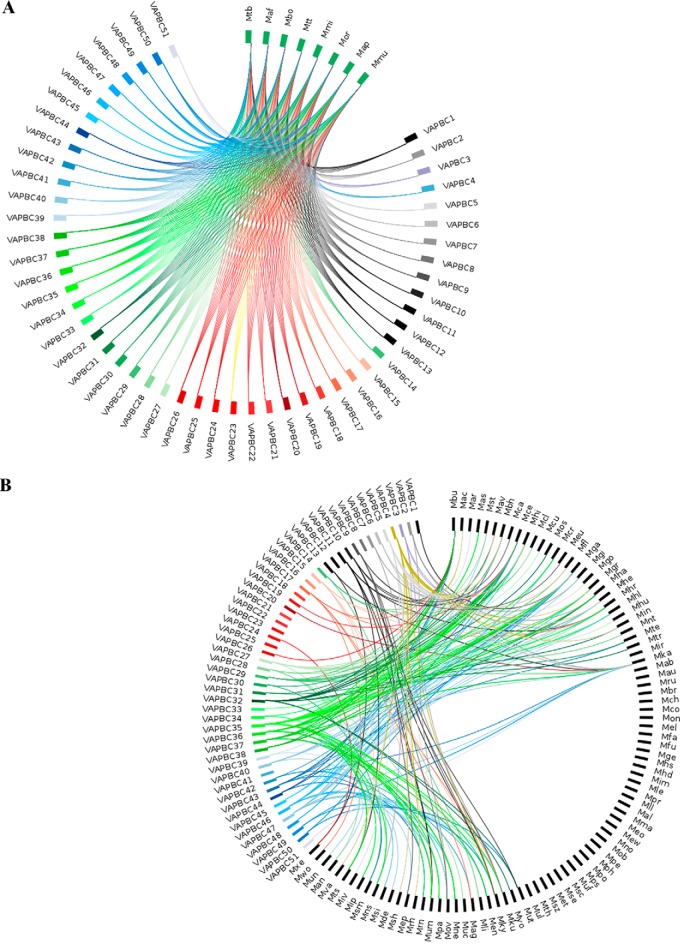
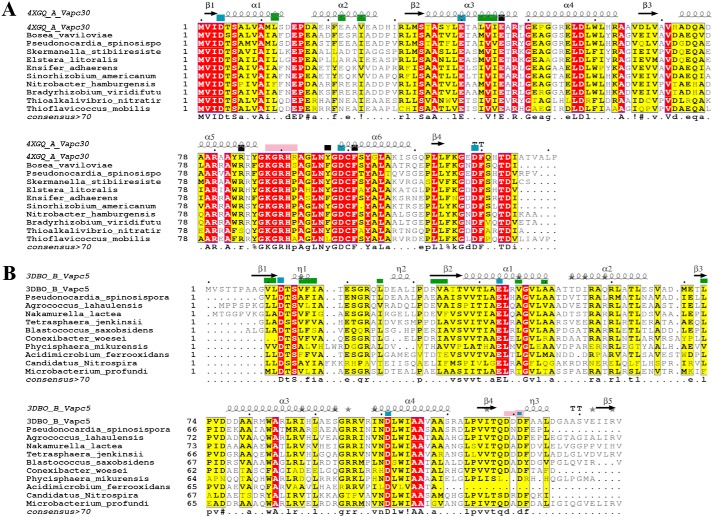
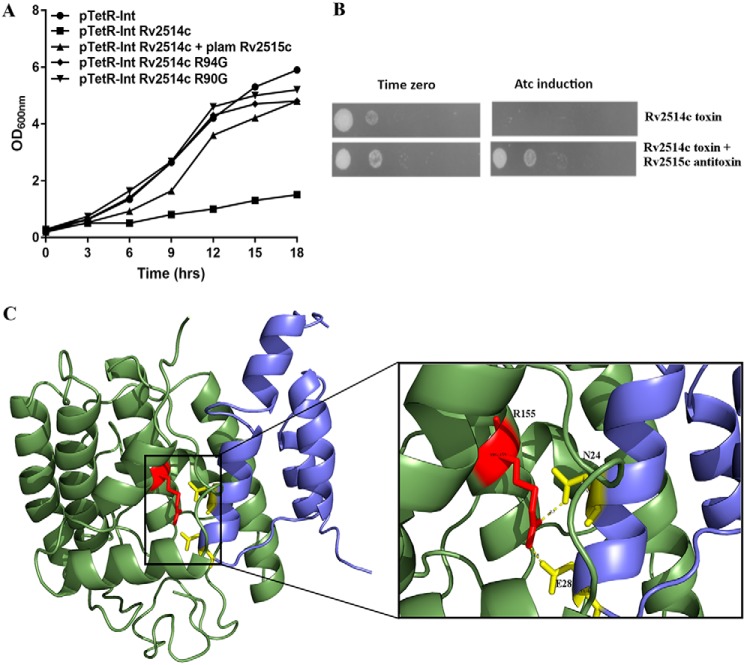
Mycobacterium tuberculosis possesses an unusually large representation of type II toxin-antitoxin (TA) systems, whose functions and targets are mostly unknown. To better understand the basis of their unique expansion and to probe putative functional similarities among these systems, here we computationally and experimentally investigated their sequence relationships. Bioinformatic and phylogenetic investigations revealed that 51 sequences of the VapBC toxin family group into paralogous sub-clusters. On the basis of conserved sequence fingerprints within paralogues, we predicted functional residues and residues at the putative TA interface that are useful to evaluate TA interactions. Substitution of these likely functional residues abolished the toxin's growth-inhibitory activity. Furthermore, conducting similarity searches in 101 mycobacterial and ∼4500 other prokaryotic genomes, we assessed the relative conservation of the M. tuberculosis TA systems and found that most TA orthologues are well-conserved among the members of the M. tuberculosis complex, which cause tuberculosis in animal hosts. We found that soil-inhabiting, free-living Actinobacteria also harbor as many as 12 TA pairs. Finally, we identified five novel putative TA modules in M. tuberculosis. For one of them, we demonstrate that overexpression of the putative toxin, Rv2514c, induces bacteriostasis and that co-expression of the cognate antitoxin Rv2515c restores bacterial growth. Taken together, our findings reveal that toxin sequences are more closely related than antitoxin sequences in M. tuberculosis Furthermore, the identification of additional TA systems reported here expands the known repertoire of TA systems in M. tuberculosis.
Keywords: Mycobacterium tuberculosis; VapBC; bioinformatics; genome analysis; homology; molecular modeling; pathogenesis; phylogenetics; protein evolution; protein sequence; structure–function; toxin–antitoxin.
© 2019 Tandon et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article.
Figures

References
-
- World Health Organization. (2018) WHO Global Tuberculosis Report 2018, World Health Organization, Geneva, Switzerland: 10.1016/j.pharep.2017.02.021 - DOI
-
- Yamaguchi Y., and Inouye M. (2016) Stress and Environmental Regulation of Gene Expression and Adaptation in Bacteria (de Bruijn F. J., ed) pp. 97–107, John Wiley & Sons, Inc., New York
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
