

Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation
- PMID: 31019026
- PMCID: PMC9512059
- DOI: 10.1126/scitranslmed.aat6177
Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation
Abstract
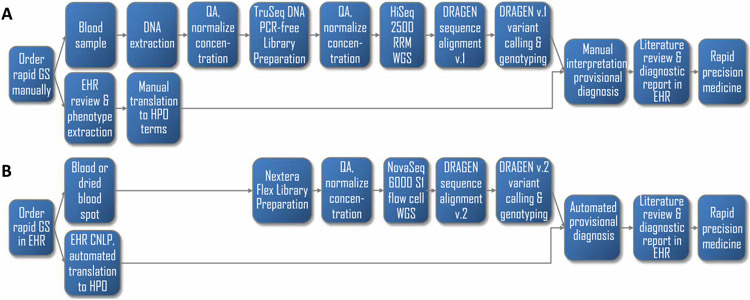
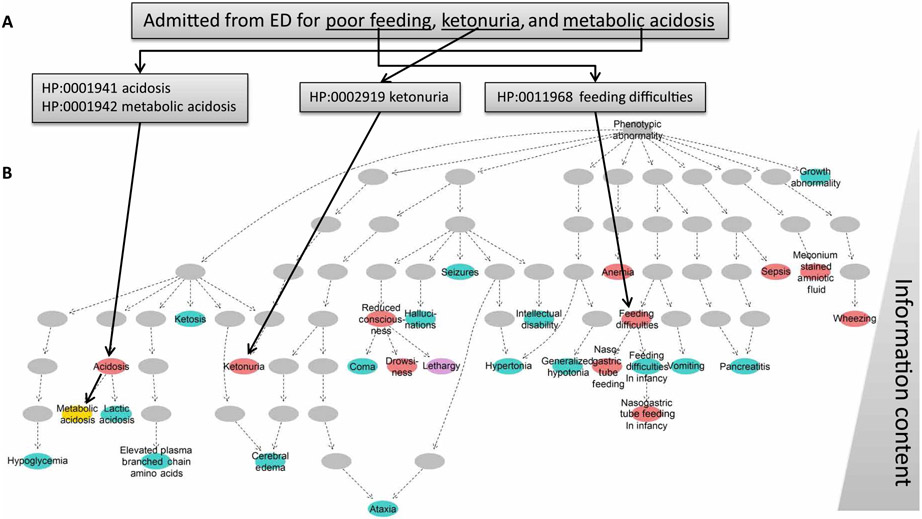
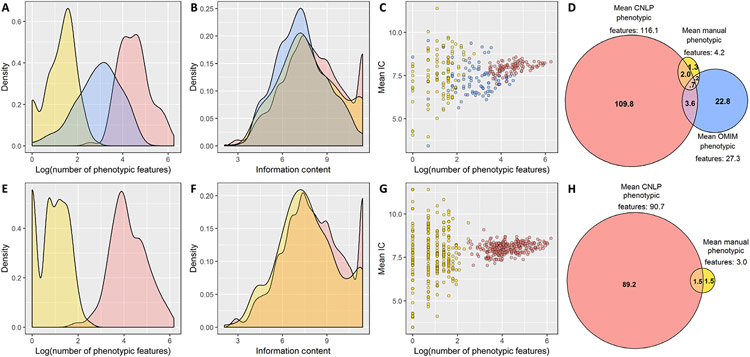
By informing timely targeted treatments, rapid whole-genome sequencing can improve the outcomes of seriously ill children with genetic diseases, particularly infants in neonatal and pediatric intensive care units (ICUs). The need for highly qualified professionals to decipher results, however, precludes widespread implementation. We describe a platform for population-scale, provisional diagnosis of genetic diseases with automated phenotyping and interpretation. Genome sequencing was expedited by bead-based genome library preparation directly from blood samples and sequencing of paired 100-nt reads in 15.5 hours. Clinical natural language processing (CNLP) automatically extracted children's deep phenomes from electronic health records with 80% precision and 93% recall. In 101 children with 105 genetic diseases, a mean of 4.3 CNLP-extracted phenotypic features matched the expected phenotypic features of those diseases, compared with a match of 0.9 phenotypic features used in manual interpretation. We automated provisional diagnosis by combining the ranking of the similarity of a patient's CNLP phenome with respect to the expected phenotypic features of all genetic diseases, together with the ranking of the pathogenicity of all of the patient's genomic variants. Automated, retrospective diagnoses concurred well with expert manual interpretation (97% recall and 99% precision in 95 children with 97 genetic diseases). Prospectively, our platform correctly diagnosed three of seven seriously ill ICU infants (100% precision and recall) with a mean time saving of 22:19 hours. In each case, the diagnosis affected treatment. Genome sequencing with automated phenotyping and interpretation in a median of 20:10 hours may increase adoption in ICUs and, thereby, timely implementation of precise treatments.
Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
Comment in
-
Rapid neonatal diagnosis.Nat Med. 2019 Jun;25(6):877. doi: 10.1038/s41591-019-0487-2. Nat Med. 2019. PMID: 31171871 No abstract available.
References
-
- Khokha MK, Mitchell LE, Wallingford JB, White paper on the study of birth defects. Birth Defects Res. 109, 180–185 (2017). - PubMed
-
- March of Dimes Foundation Data Book for Policy Makers: Maternal, Infant and Child Health in the United States 2016 (March of Dimes, 2016); www.marchofdimes.org/March-of-Dimes-2016-Databook.pdf.
-
- Murphy SL, Xu J, Kochanek KD, Arias E, Mortality in the United States, 2017. NCHS Data Brief, 1–8 (2018). - PubMed
-
- Yoon PW, Olney RS, Khoury MJ, Sappenfield WM, Chavez GF, Taylor D, Contribution of birth defects and genetic diseases to pediatric hospitalizations. A population-based study. Arch. Pediatr. Adolesc. Med 151, 1096–1103 (1997). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
