

Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity
- PMID: 31019077
- PMCID: PMC6511047
- DOI: 10.1073/pnas.1814932116
Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity
Abstract
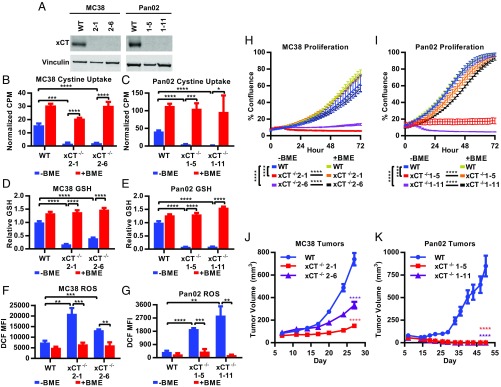
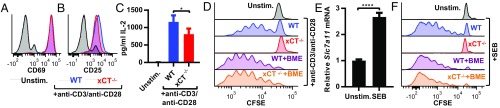
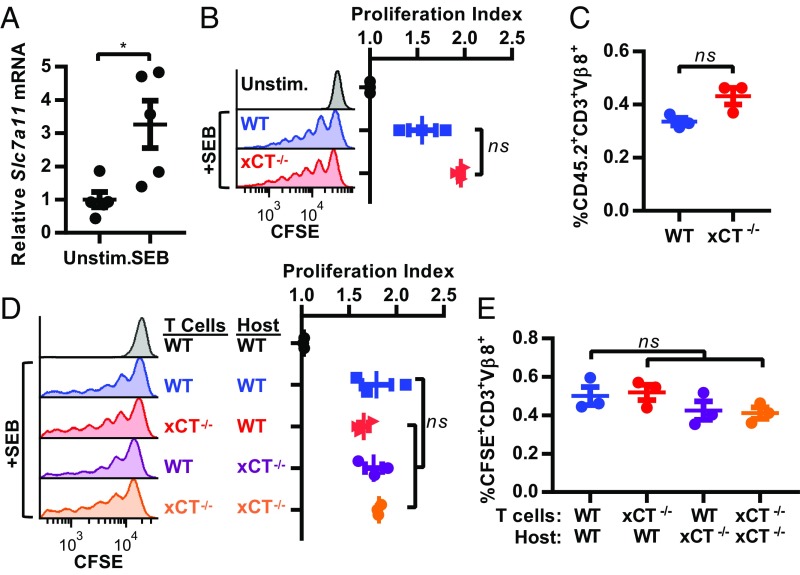
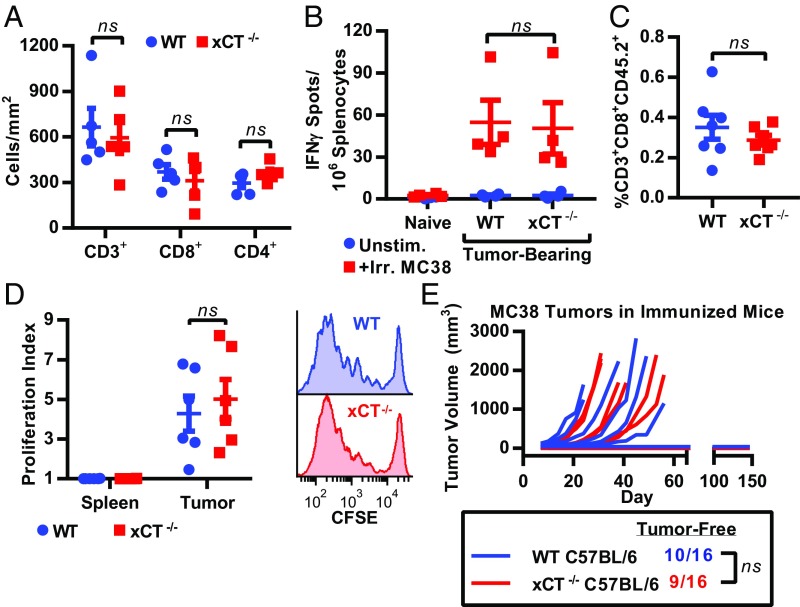
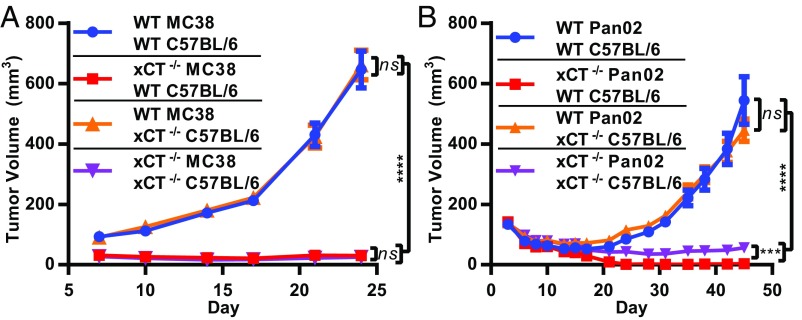
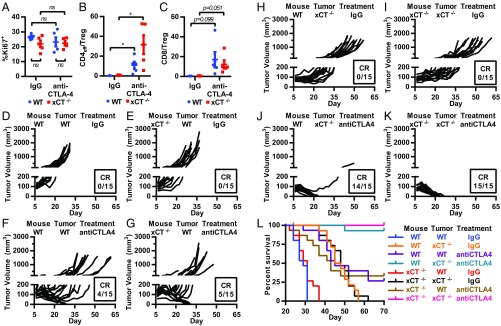
T cell-invigorating cancer immunotherapies have near-curative potential. However, their clinical benefit is currently limited, as only a fraction of patients respond, suggesting that these regimens may benefit from combination with tumor-targeting treatments. As oncogenic progression is accompanied by alterations in metabolic pathways, tumors often become heavily reliant on antioxidant machinery and may be susceptible to increases in oxidative stress. The cystine-glutamate antiporter xCT is frequently overexpressed in cancer and fuels the production of the antioxidant glutathione; thus, tumors prone to redox stress may be selectively vulnerable to xCT disruption. However, systemic inhibition of xCT may compromise antitumor immunity, as xCT is implicated in supporting antigen-induced T cell proliferation. Therefore, we utilized immune-competent murine tumor models to investigate whether cancer cell expression of xCT was required for tumor growth in vivo and if deletion of host xCT impacted antitumor immune responses. Deletion of xCT in tumor cells led to defective cystine uptake, accumulation of reactive oxygen species, and impaired tumor growth, supporting a cancer cell-autonomous role for xCT. In contrast, we observed that, although T cell proliferation in culture was exquisitely dependent on xCT expression, xCT was dispensable for T cell proliferation in vivo and for the generation of primary and memory immune responses to tumors. These findings prompted the combination of tumor cell xCT deletion with the immunotherapeutic agent anti-CTLA-4, which dramatically increased the frequency and durability of antitumor responses. Together, these results identify a metabolic vulnerability specific to tumors and demonstrate that xCT disruption can expand the efficacy of anticancer immunotherapies.
Keywords: T cells; cancer; cystine; immunotherapy; xCT.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
Conflict of interest statement: All authors are employees and shareholders of Pfizer.
Figures
References
-
- Reczek CR, Chandel NS. The two faces of reactive oxygen species in cancer. Annu Rev Cancer Biol. 2017;1:79–98.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
