

A comparative analysis of methods for de novo assembly of hymenopteran genomes using either haploid or diploid samples
- PMID: 31019201
- PMCID: PMC6482151
- DOI: 10.1038/s41598-019-42795-6
A comparative analysis of methods for de novo assembly of hymenopteran genomes using either haploid or diploid samples
Abstract
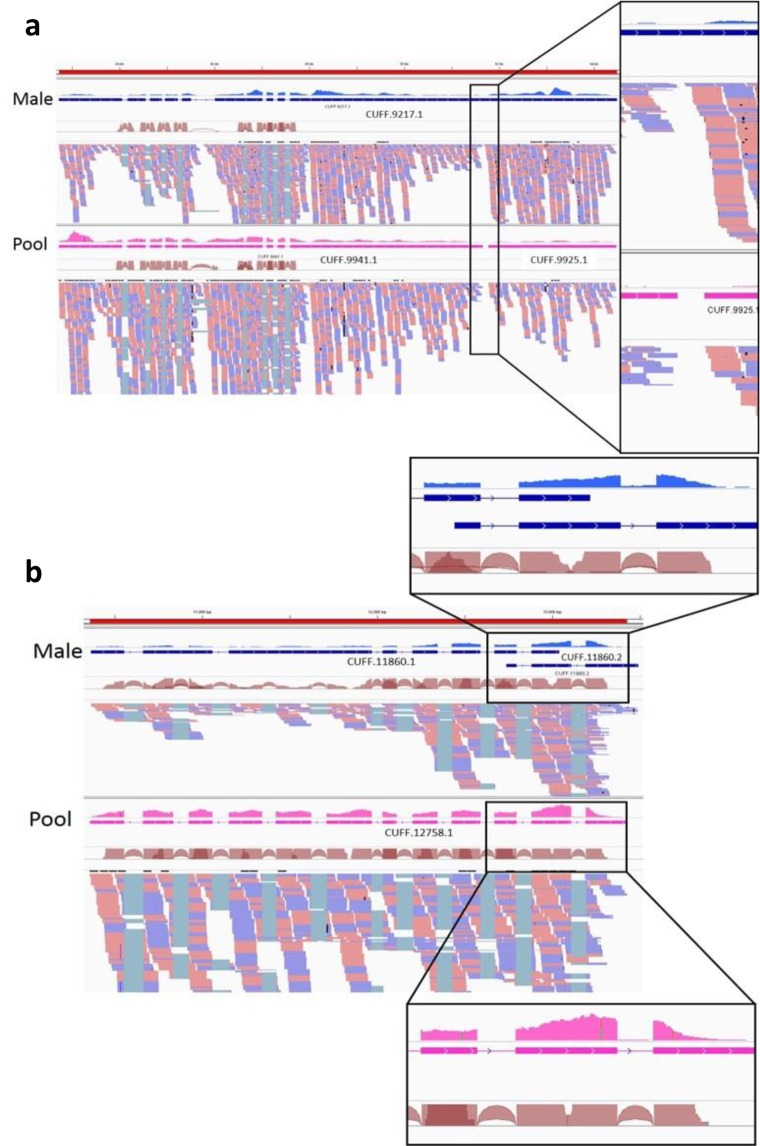
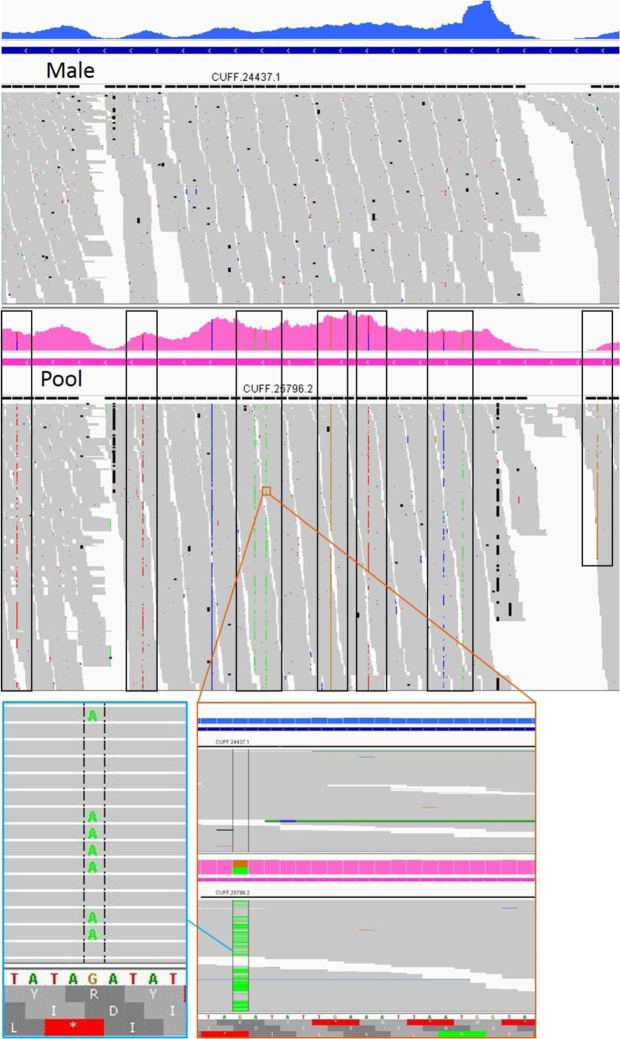
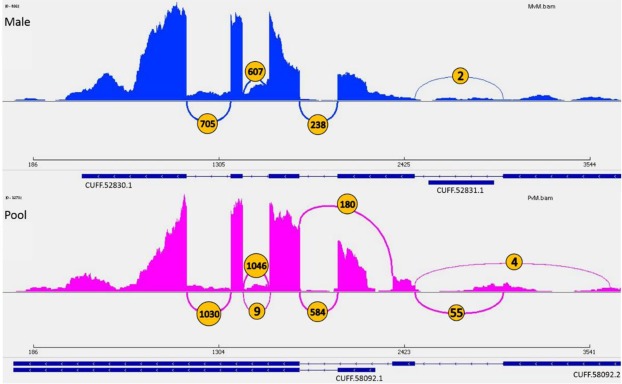
Diverse invertebrate taxa including all 200,000 species of Hymenoptera (ants, bees, wasps and sawflies) have a haplodiploid sex determination system, where females are diploid and males are haploid. Thus, hymenopteran genome projects can make use of DNA from a single haploid male sample, which is assumed advantageous for genome assembly. For the purpose of gene annotation, transcriptome sequencing is usually conducted using RNA from a pool of individuals. We conducted a comparative analysis of genome and transcriptome assembly and annotation methods, using genetic sources of different ploidy: (1) DNA from a haploid male or a diploid female (2) RNA from the same haploid male or a pool of individuals. We predicted that the use of a haploid male as opposed to a diploid female will simplify the genome assembly and gene annotation thanks to the lack of heterozygosity. Using DNA and RNA from the same haploid individual is expected to provide better confidence in transcript-to-genome alignment, and improve the annotation of gene structure in terms of the exon/intron boundaries. The haploid genome assemblies proved to be more contiguous, with both contig and scaffold N50 size at least threefold greater than their diploid counterparts. Completeness evaluation showed mixed results. The SOAPdenovo2 diploid assembly was missing more genes than the haploid assembly. The SPAdes diploid assembly had more complete genes, but a higher level of duplicates, and a greatly overestimated genome size. When aligning the two transcriptomes against the male genome, the male transcriptome gave 2-3% more complete transcripts than the pool transcriptome for genes with comparable expression levels in both transcriptomes. However, this advantage disappears in the final results of the gene annotation pipeline that incorporates evidence from homologous proteins. The RNA pool is still required to obtain the full transcriptome with genes that are expressed in other life stages and castes. In conclusion, the use of a haploid source material for a de novo genome project provides a substantial advantage to the quality of the genome draft and the use of RNA from the same haploid individual for transcriptome to genome alignment provides a minor advantage for genes that are expressed in the adult male.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
