

NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling
- PMID: 31019297
- PMCID: PMC7138021
- DOI: 10.1038/s41586-019-1150-2
NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling
Abstract
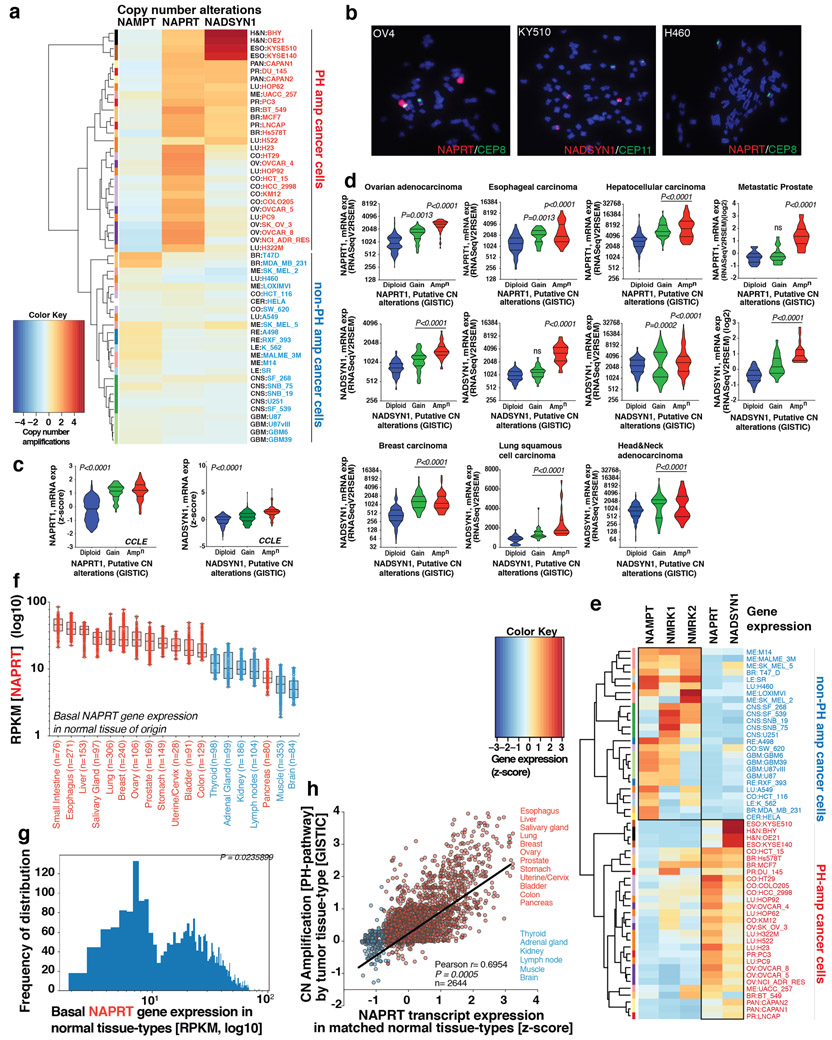
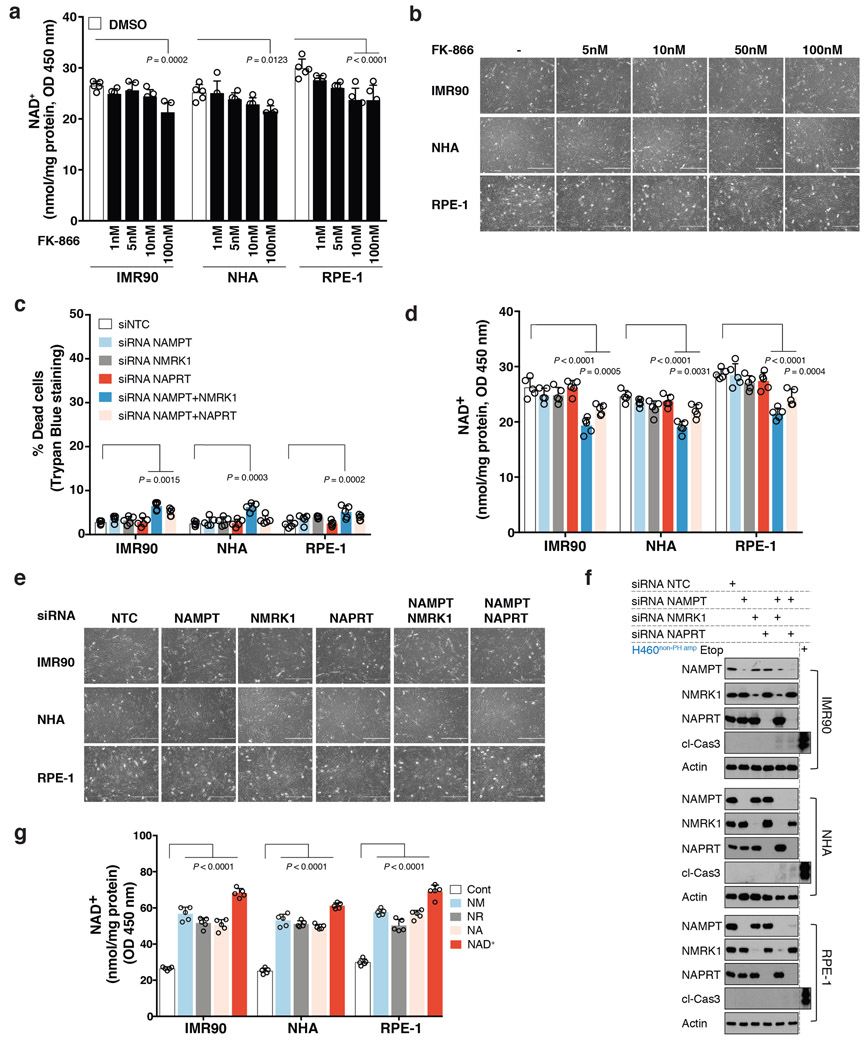
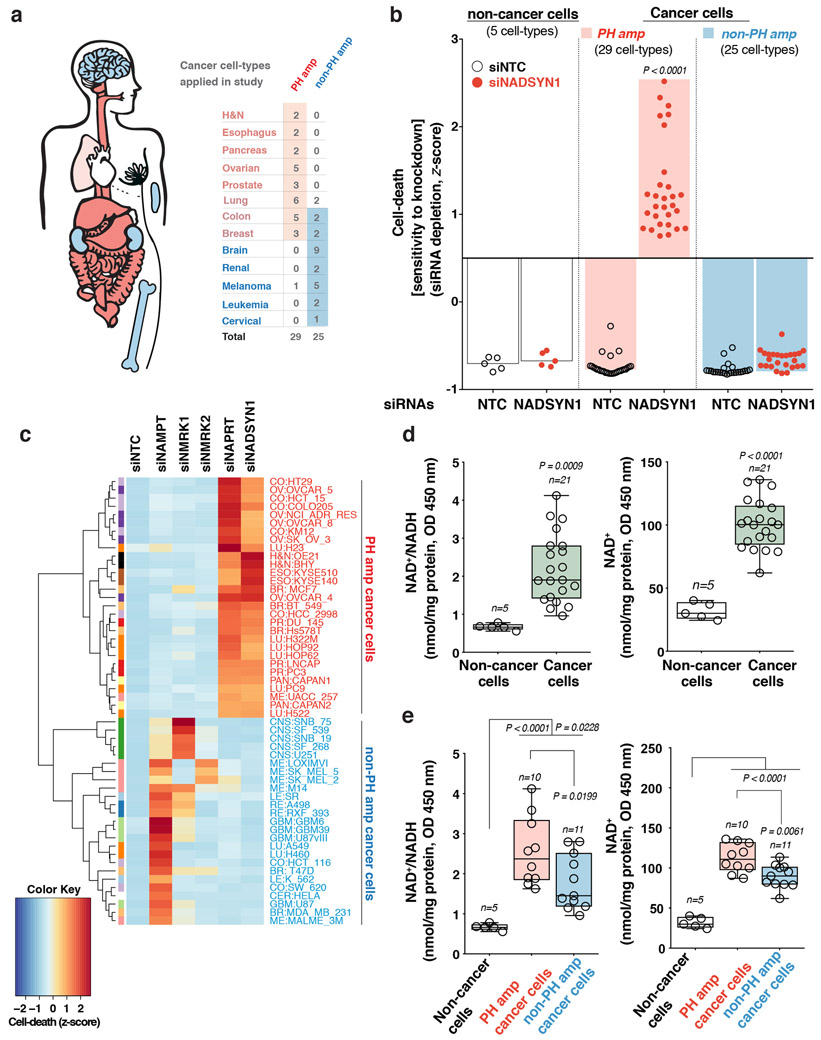
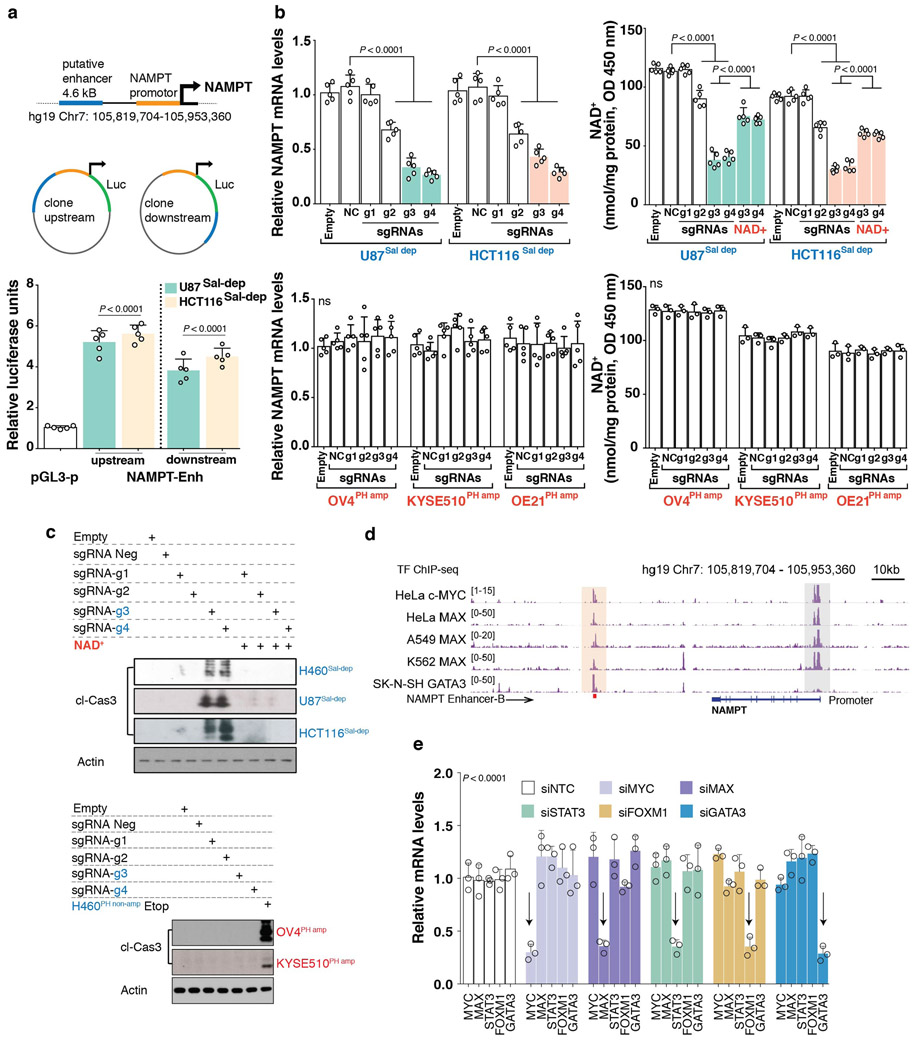
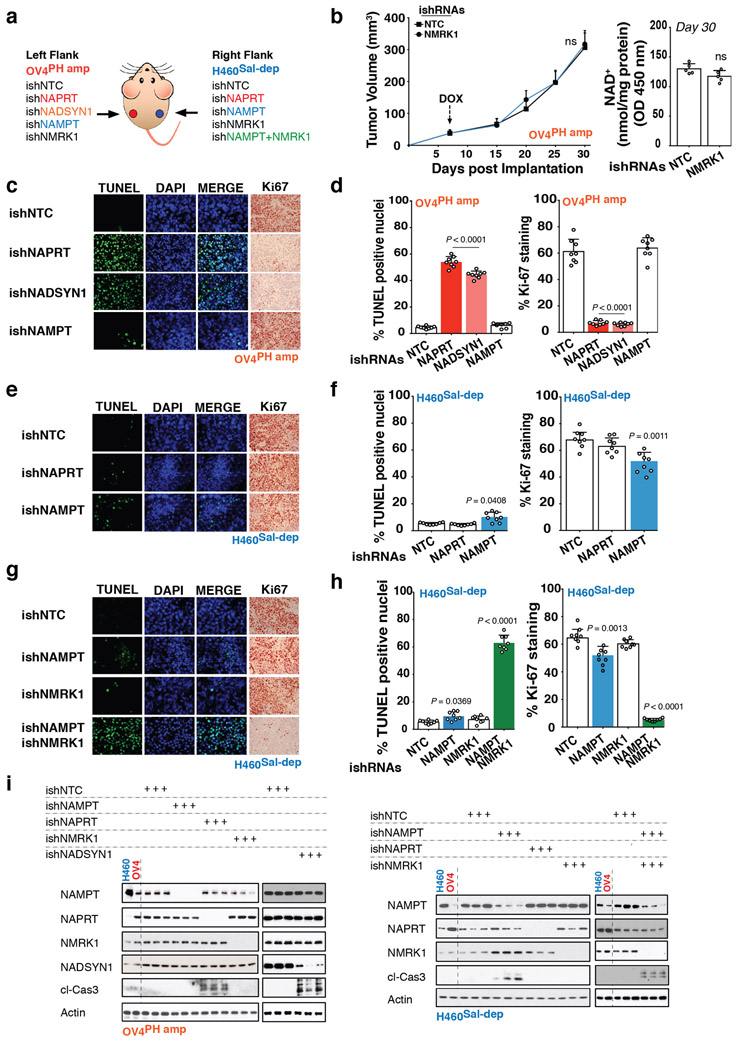
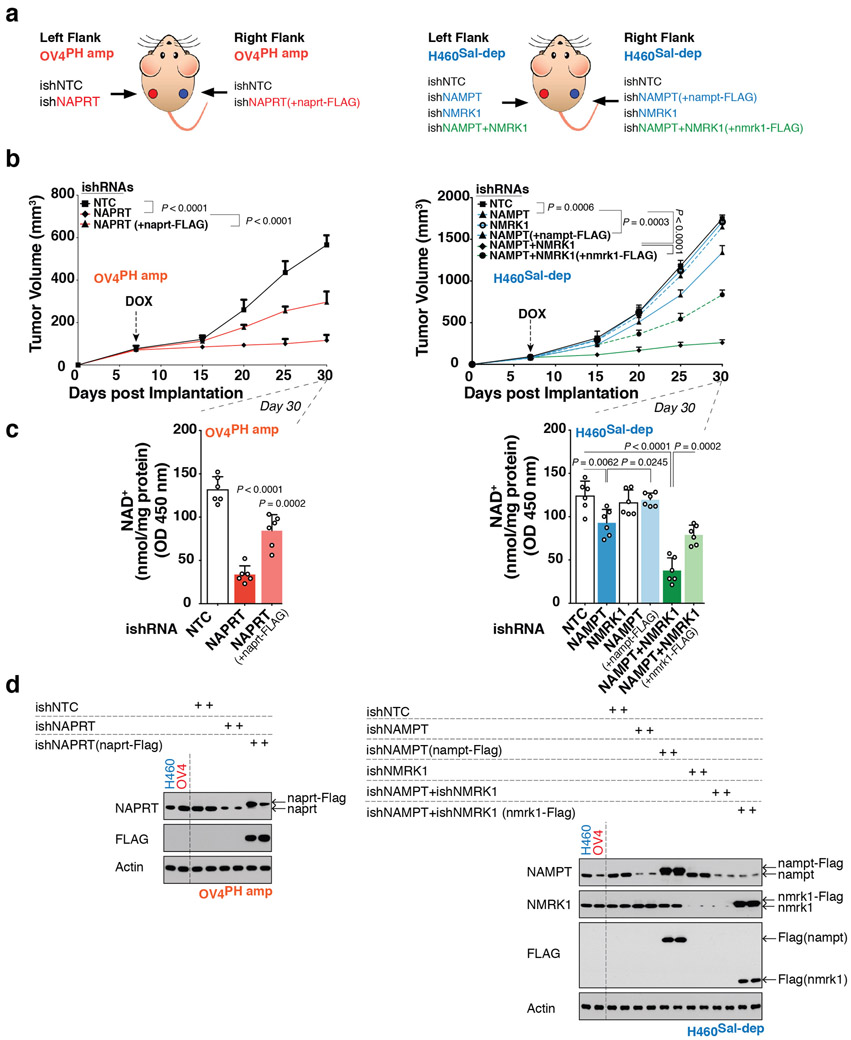
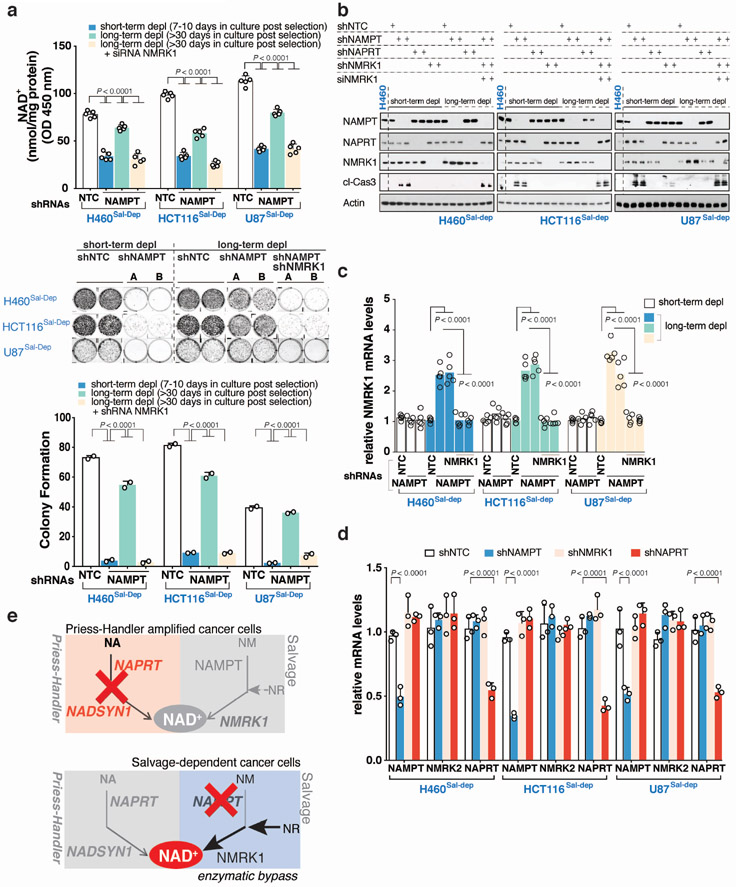
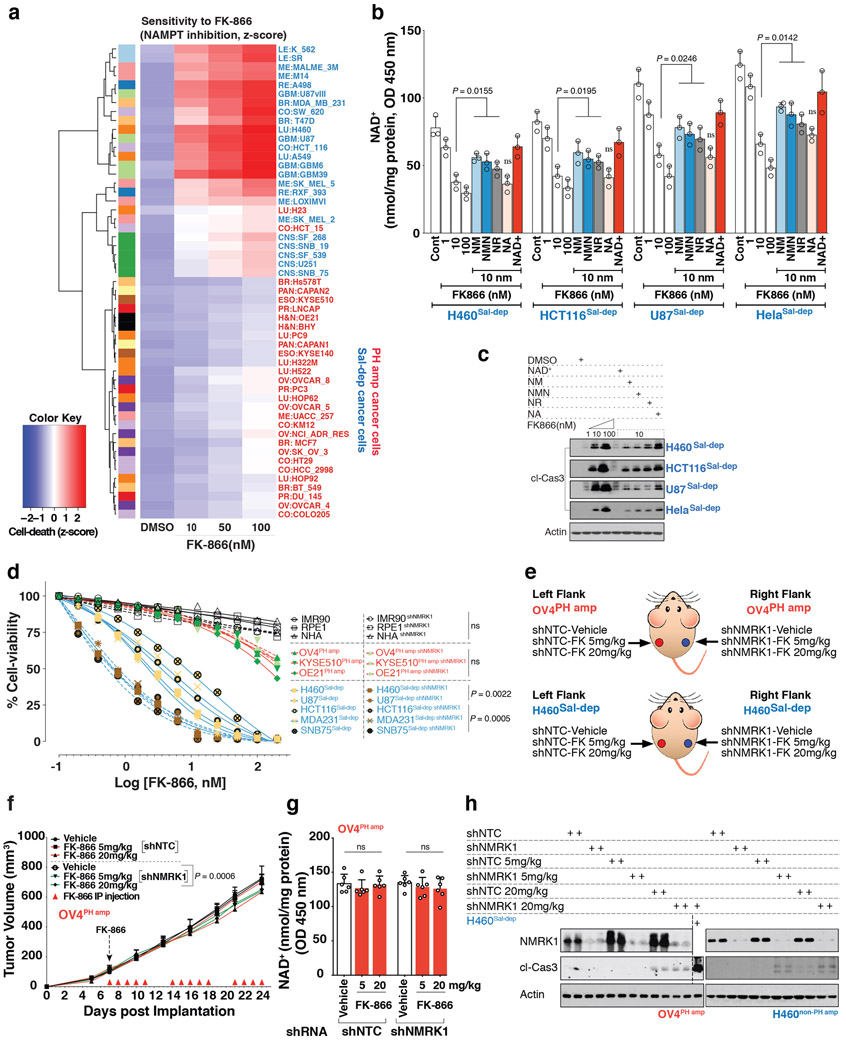
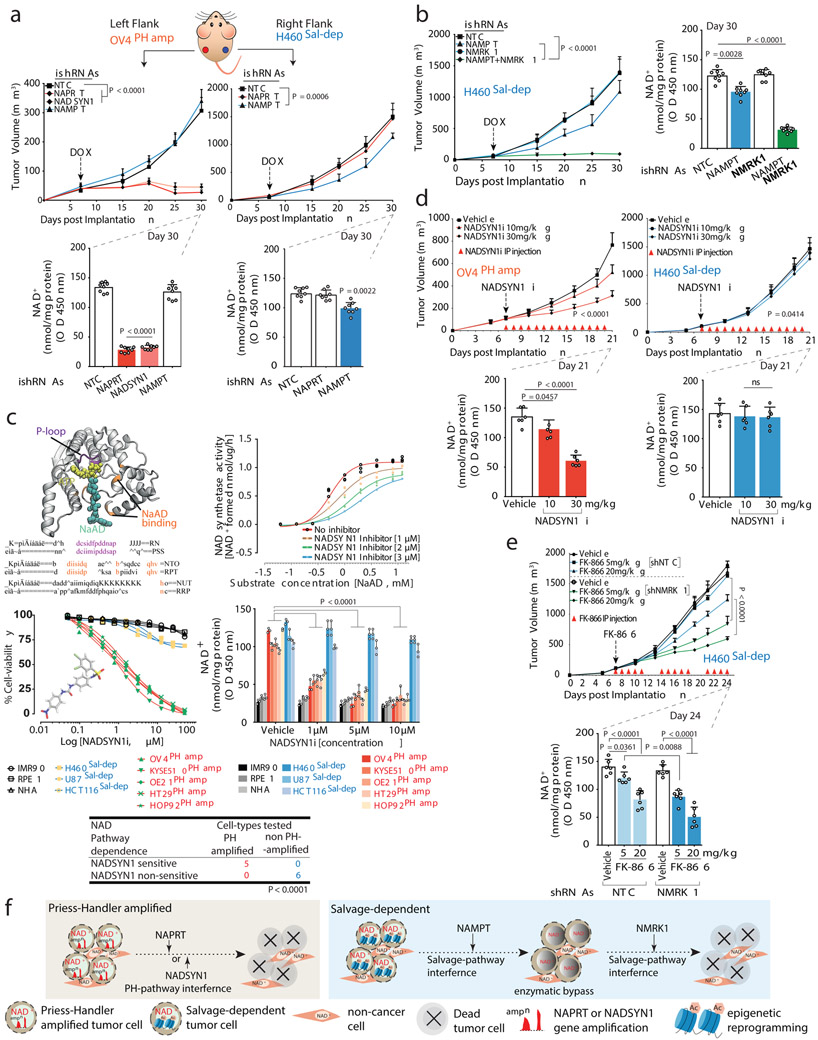
Precision oncology hinges on linking tumour genotype with molecularly targeted drugs1; however, targeting the frequently dysregulated metabolic landscape of cancer has proven to be a major challenge2. Here we show that tissue context is the major determinant of dependence on the nicotinamide adenine dinucleotide (NAD) metabolic pathway in cancer. By analysing more than 7,000 tumours and 2,600 matched normal samples of 19 tissue types, coupled with mathematical modelling and extensive in vitro and in vivo analyses, we identify a simple and actionable set of 'rules'. If the rate-limiting enzyme of de novo NAD synthesis, NAPRT, is highly expressed in a normal tissue type, cancers that arise from that tissue will have a high frequency of NAPRT amplification and be completely and irreversibly dependent on NAPRT for survival. By contrast, tumours that arise from normal tissues that do not express NAPRT highly are entirely dependent on the NAD salvage pathway for survival. We identify the previously unknown enhancer that underlies this dependence. Amplification of NAPRT is shown to generate a pharmacologically actionable tumour cell dependence for survival. Dependence on another rate-limiting enzyme of the NAD synthesis pathway, NAMPT, as a result of enhancer remodelling is subject to resistance by NMRK1-dependent synthesis of NAD. These results identify a central role for tissue context in determining the choice of NAD biosynthetic pathway, explain the failure of NAMPT inhibitors, and pave the way for more effective treatments.
Figures

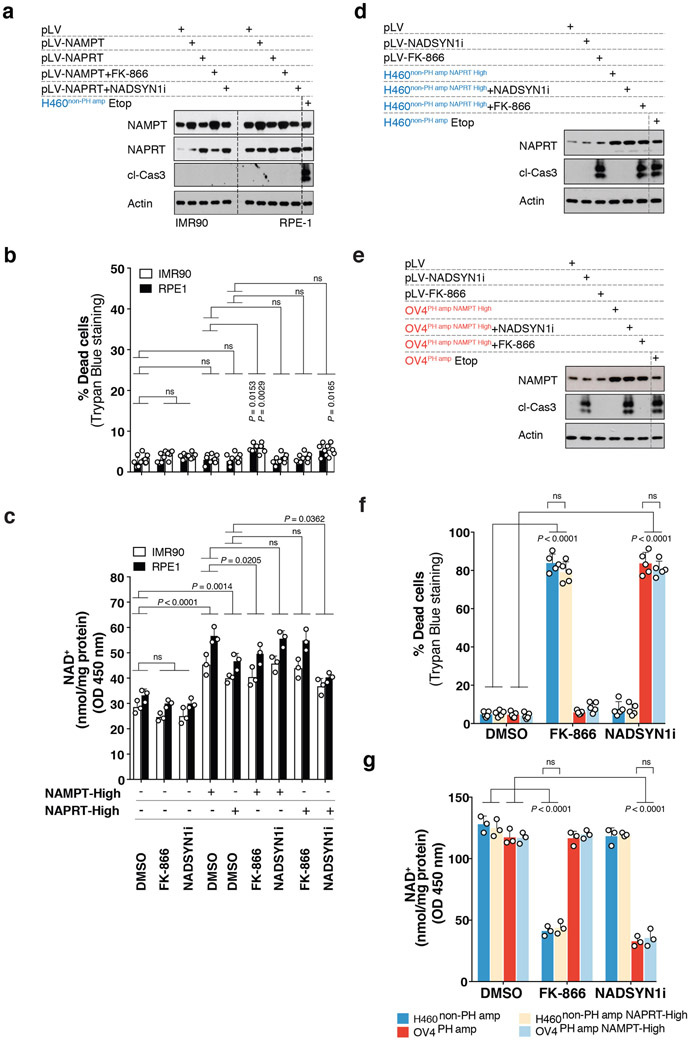
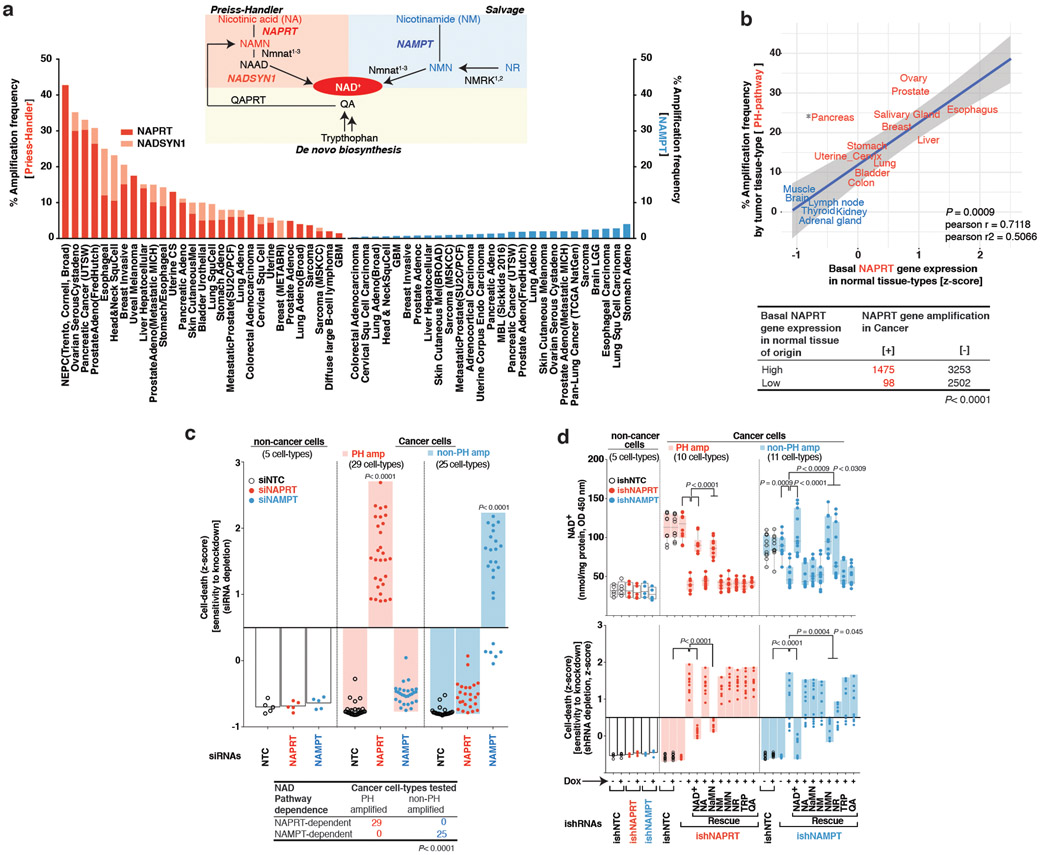
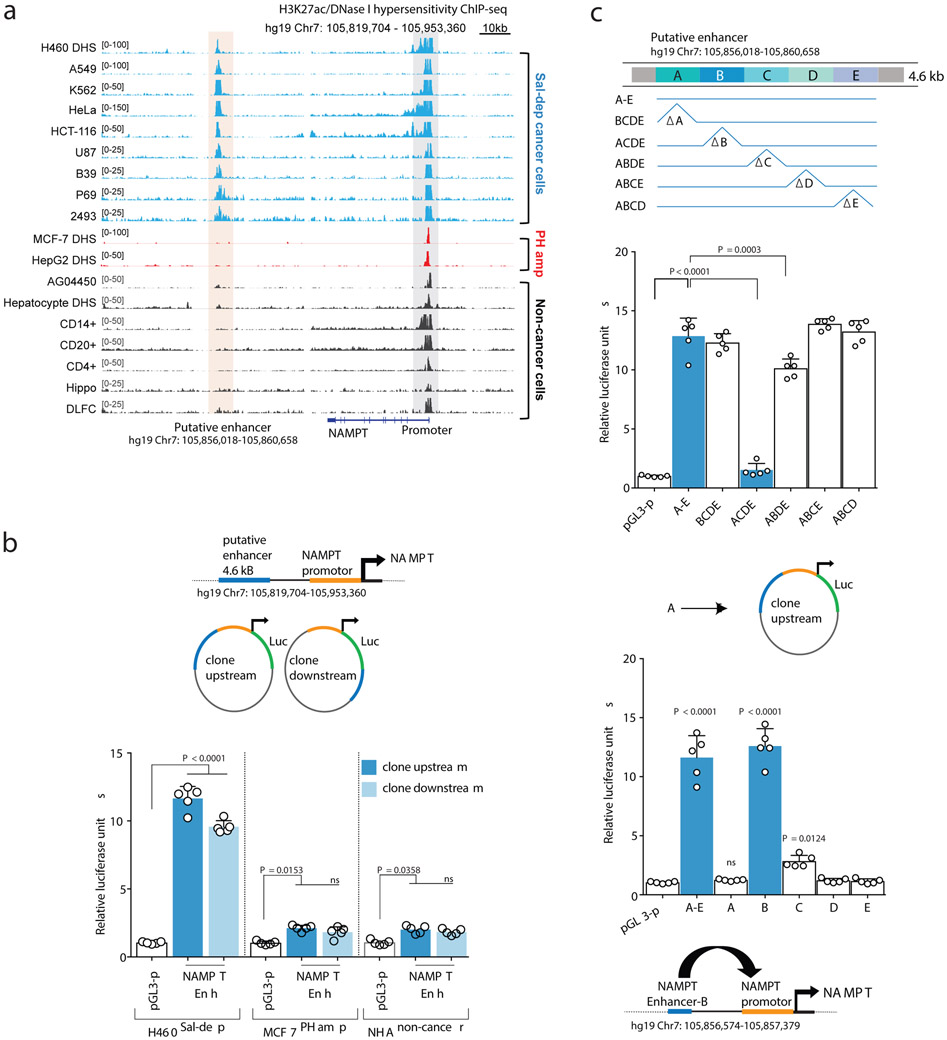
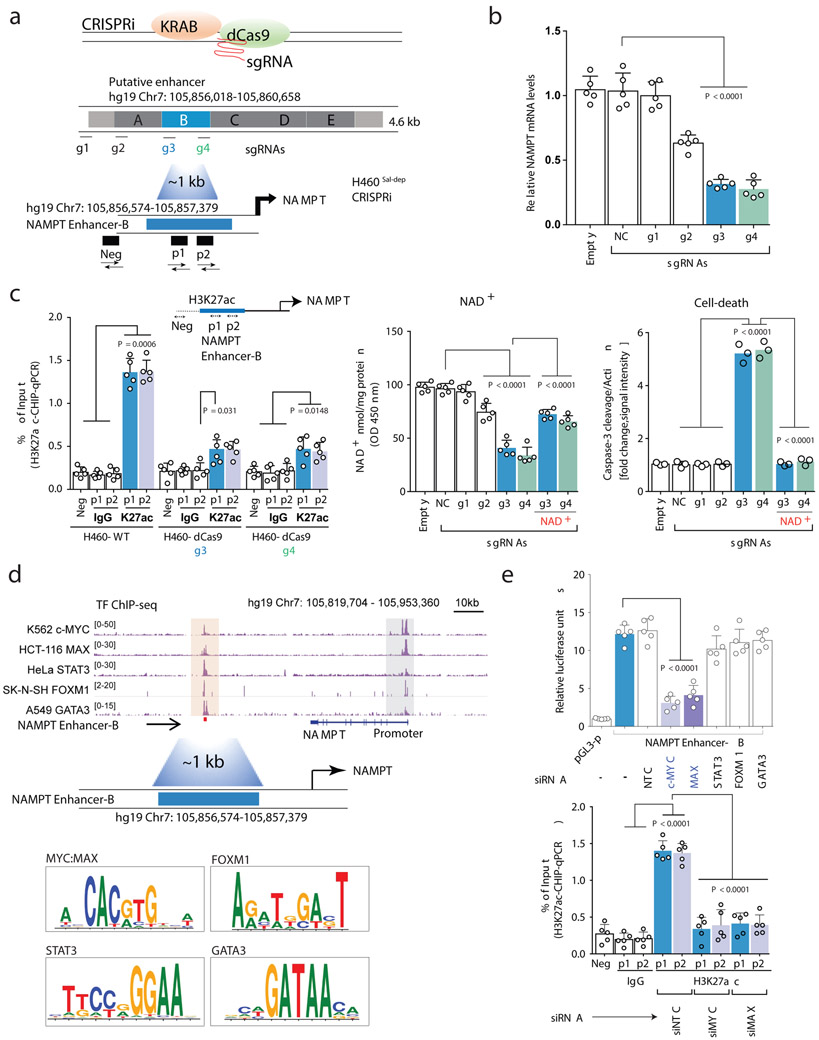
Comment in
-
Context determines which pathway to use for NAD synthesis.Nat Rev Cancer. 2019 Jul;19(7):365. doi: 10.1038/s41568-019-0157-1. Nat Rev Cancer. 2019. PMID: 31138903 No abstract available.
-
Slide-Seq for Spatially Mapping Gene Expression. Metabolic Syndrome Exacerbates Group 2 Pulmonary Hypertension, and NAD Metabolism Is Influenced by Tissue Origin.Am J Respir Cell Mol Biol. 2020 Jan;62(1):112-114. doi: 10.1165/rcmb.2019-0333RO. Am J Respir Cell Mol Biol. 2020. PMID: 31633380 No abstract available.
References
-
- Roychowdhury S and Chinnaiyan AM. Translating genomics for precision cancer medicine. Annu Rev Genomics Hum Genet. 15: 395–415. 2014. - PubMed
-
- Mayers JR and Vander Heiden MG. Nature and Nurture: What Determines Tumor Metabolic Phenotypes. Cancer Res. 77(12): 3131–4. 2017. - PubMed
-
- Verdin E NAD+ in aging, metabolism, and neurodegeneration. Science. 350(6265):1208–1213. 2015. - PubMed
Supplementary References
-
- Heintzman ND. et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 39: 311–318. 2007. - PubMed
-
- Hartigan JA; Hartigan PM The Dip Test of Unimodality. Ann. Statist. 13 (1):70–84. 1985.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
