

Proteogenomic Annotation of Chinese Hamsters Reveals Extensive Novel Translation Events and Endogenous Retroviral Elements
- PMID: 31020842
- PMCID: PMC6571120
- DOI: 10.1021/acs.jproteome.8b00935
Proteogenomic Annotation of Chinese Hamsters Reveals Extensive Novel Translation Events and Endogenous Retroviral Elements
Abstract
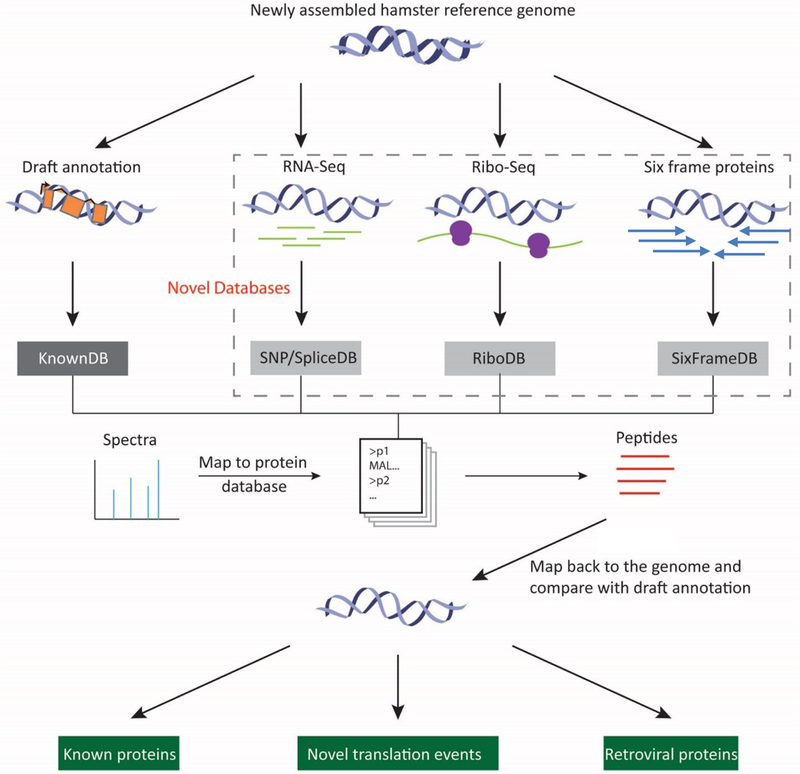
A high-quality genome annotation greatly facilitates successful cell line engineering. Standard draft genome annotation pipelines are based largely on de novo gene prediction, homology, and RNA-Seq data. However, draft annotations can suffer from incorrect predictions of translated sequence, inaccurate splice isoforms, and missing genes. Here, we generated a draft annotation for the newly assembled Chinese hamster genome and used RNA-Seq, proteomics, and Ribo-Seq to experimentally annotate the genome. We identified 3529 new proteins compared to the hamster RefSeq protein annotation and 2256 novel translational events (e.g., alternative splices, mutations, and novel splices). Finally, we used this pipeline to identify the source of translated retroviruses contaminating recombinant products from Chinese hamster ovary (CHO) cell lines, including 119 type-C retroviruses, thus enabling future efforts to eliminate retroviruses to reduce the costs incurred with retroviral particle clearance. In summary, the improved annotation provides a more accurate resource for CHO cell line engineering, by facilitating the interpretation of omics data, defining of cellular pathways, and engineering of complex phenotypes.
Keywords: Chinese hamster; endogenous retrovirus; genome annotation; proteogenomics.
Figures




References
-
- Brinkrolf K; Rupp O; Laux H; Kollin F; Ernst W; Linke B; Kofler R; Romand S; Hesse F; Budach WE; et al. Chinese Hamster Genome Sequenced from Sorted Chromosomes. Nat. Biotechnol. 2013, 31, 694. - PubMed
