

Mutation update for the SATB2 gene
- PMID: 31021519
- PMCID: PMC11431158
- DOI: 10.1002/humu.23771
Mutation update for the SATB2 gene
Abstract
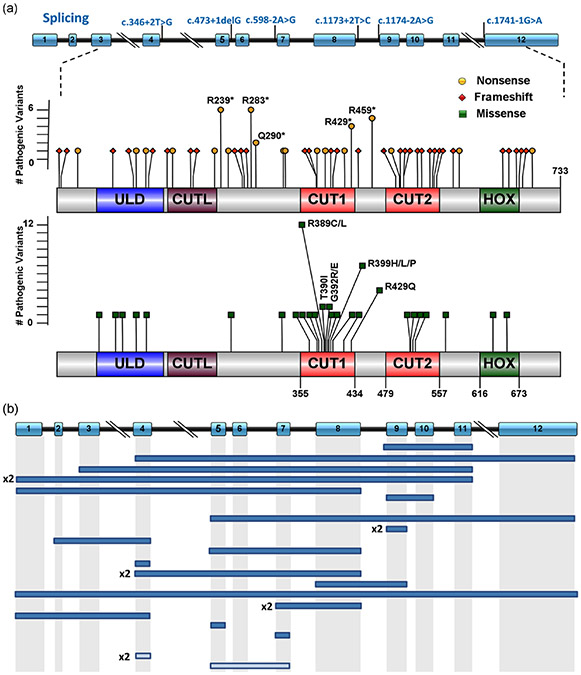
SATB2-associated syndrome (SAS) is an autosomal dominant neurodevelopmental disorder caused by alterations in the SATB2 gene. Here we present a review of published pathogenic variants in the SATB2 gene to date and report 38 novel alterations found in 57 additional previously unreported individuals. Overall, we present a compilation of 120 unique variants identified in 155 unrelated families ranging from single nucleotide coding variants to genomic rearrangements distributed throughout the entire coding region of SATB2. Single nucleotide variants predicted to result in the occurrence of a premature stop codon were the most commonly seen (51/120 = 42.5%) followed by missense variants (31/120 = 25.8%). We review the rather limited functional characterization of pathogenic variants and discuss current understanding of the consequences of the different molecular alterations. We present an expansive phenotypic review along with novel genotype-phenotype correlations. Lastly, we discuss current knowledge of animal models and present future prospects. This review should help provide better guidance for the care of individuals diagnosed with SAS.
Keywords: SATB2; SATB2-associated syndrome; genotype-phenotype correlation; pathogenic variants; whole exome sequencing.
© 2019 Wiley Periodicals, Inc.
Conflict of interest statement
CONFLICT OF INTERESTS
The authors declare that there is no conflict of interests.
Figures

References
-
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Farinas I, Grosschedl R, & McConnell SK (2008). Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron, 57(3), 364–377. - PubMed
-
- Balasubramanian M, Smith K, Basel-Vanagaite L, Feingold MF, Brock P, Gowans GC, & Parker MJ (2011). Case series: 2q33.1 microdeletion syndrome--further delineation of the phenotype. Journal of Medical Genetics, 48(5), 290–298. - PubMed
