

Genome-wide sexually antagonistic variants reveal long-standing constraints on sexual dimorphism in fruit flies
- PMID: 31022179
- PMCID: PMC6504117
- DOI: 10.1371/journal.pbio.3000244
Genome-wide sexually antagonistic variants reveal long-standing constraints on sexual dimorphism in fruit flies
Abstract
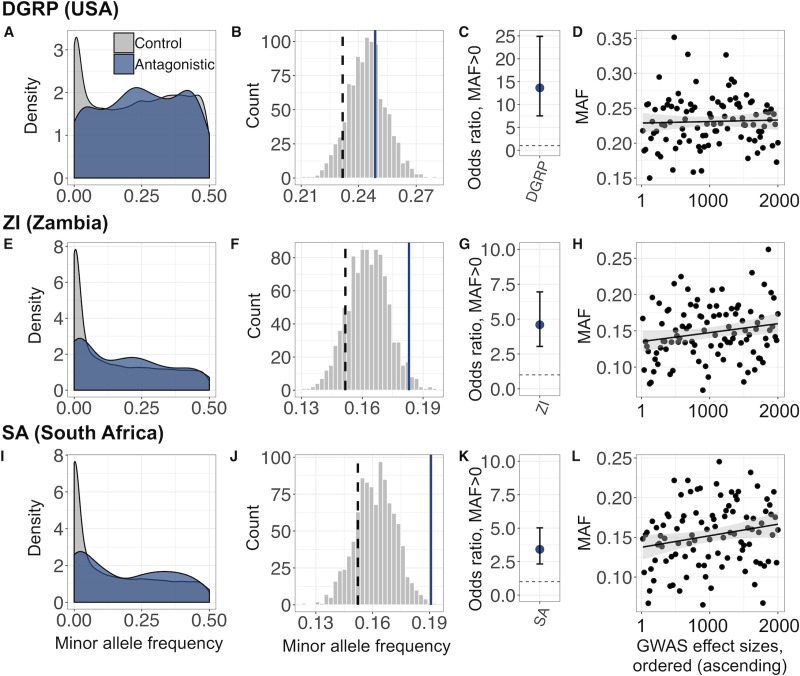
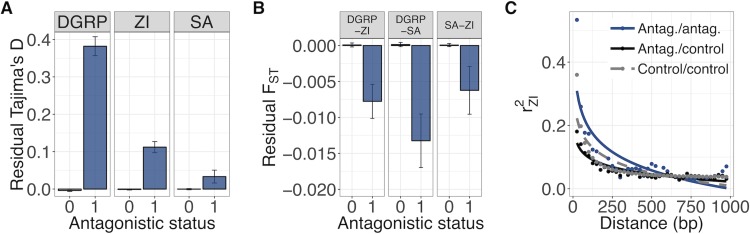
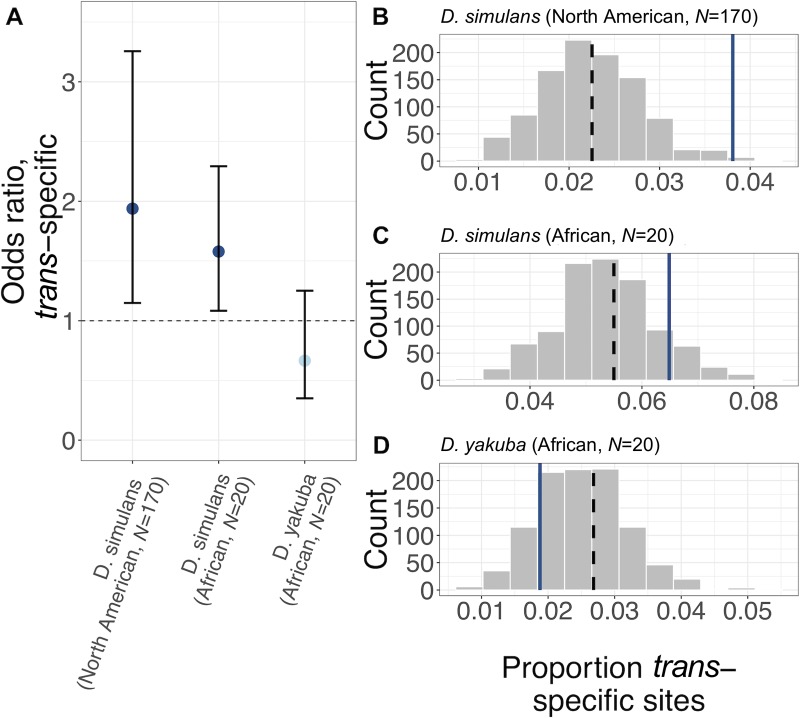
The evolution of sexual dimorphism is constrained by a shared genome, leading to 'sexual antagonism', in which different alleles at given loci are favoured by selection in males and females. Despite its wide taxonomic incidence, we know little about the identity, genomic location, and evolutionary dynamics of antagonistic genetic variants. To address these deficits, we use sex-specific fitness data from 202 fully sequenced hemiclonal Drosophila melanogaster fly lines to perform a genome-wide association study (GWAS) of sexual antagonism. We identify approximately 230 chromosomal clusters of candidate antagonistic single nucleotide polymorphisms (SNPs). In contradiction to classic theory, we find no clear evidence that the X chromosome is a hot spot for sexually antagonistic variation. Characterising antagonistic SNPs functionally, we find a large excess of missense variants but little enrichment in terms of gene function. We also assess the evolutionary persistence of antagonistic variants by examining extant polymorphism in wild D. melanogaster populations and closely related species. Remarkably, antagonistic variants are associated with multiple signatures of balancing selection across the D. melanogaster distribution range and in their sister species D. simulans, indicating widespread and evolutionarily persistent (about 1 million years) genomic constraints on the evolution of sexual dimorphism. Based on our results, we propose that antagonistic variation accumulates because of constraints on the resolution of sexual conflict over protein coding sequences, thus contributing to the long-term maintenance of heritable fitness variation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
