

Performance Assessment and Selection of Normalization Procedures for Single-Cell RNA-Seq
- PMID: 31022373
- PMCID: PMC6544759
- DOI: 10.1016/j.cels.2019.03.010
Performance Assessment and Selection of Normalization Procedures for Single-Cell RNA-Seq
Abstract
Systematic measurement biases make normalization an essential step in single-cell RNA sequencing (scRNA-seq) analysis. There may be multiple competing considerations behind the assessment of normalization performance, of which some may be study specific. We have developed "scone"- a flexible framework for assessing performance based on a comprehensive panel of data-driven metrics. Through graphical summaries and quantitative reports, scone summarizes trade-offs and ranks large numbers of normalization methods by panel performance. The method is implemented in the open-source Bioconductor R software package scone. We show that top-performing normalization methods lead to better agreement with independent validation data for a collection of scRNA-seq datasets. scone can be downloaded at http://bioconductor.org/packages/scone/.
Keywords: RNA-seq; methods; normalization; preprocessing; quality control; scRNA-seq; single-cell.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures

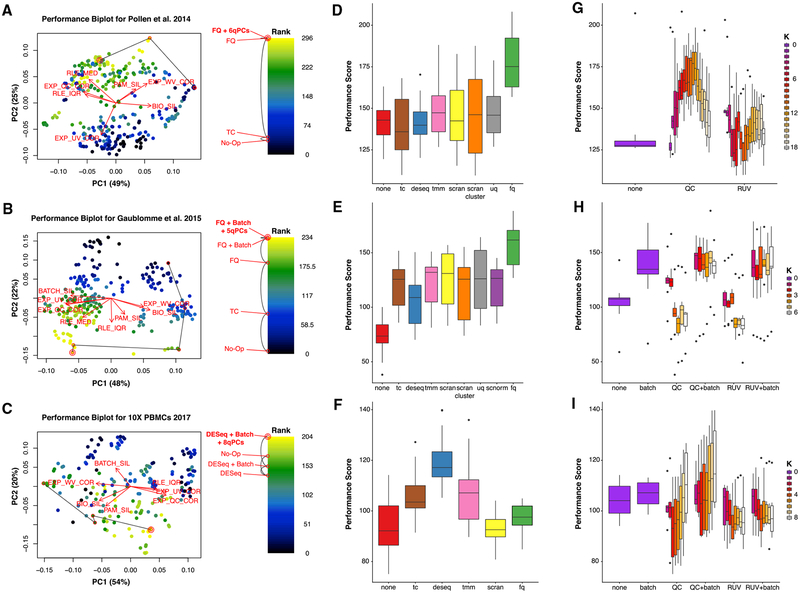
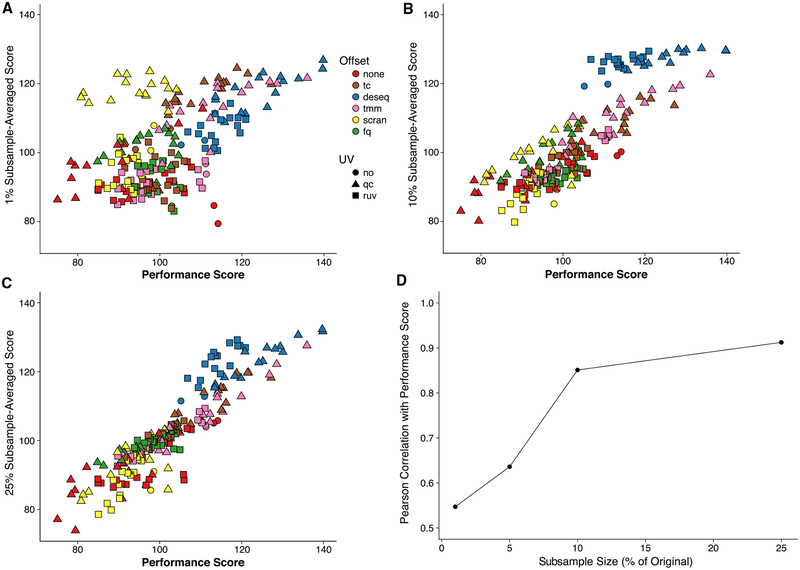
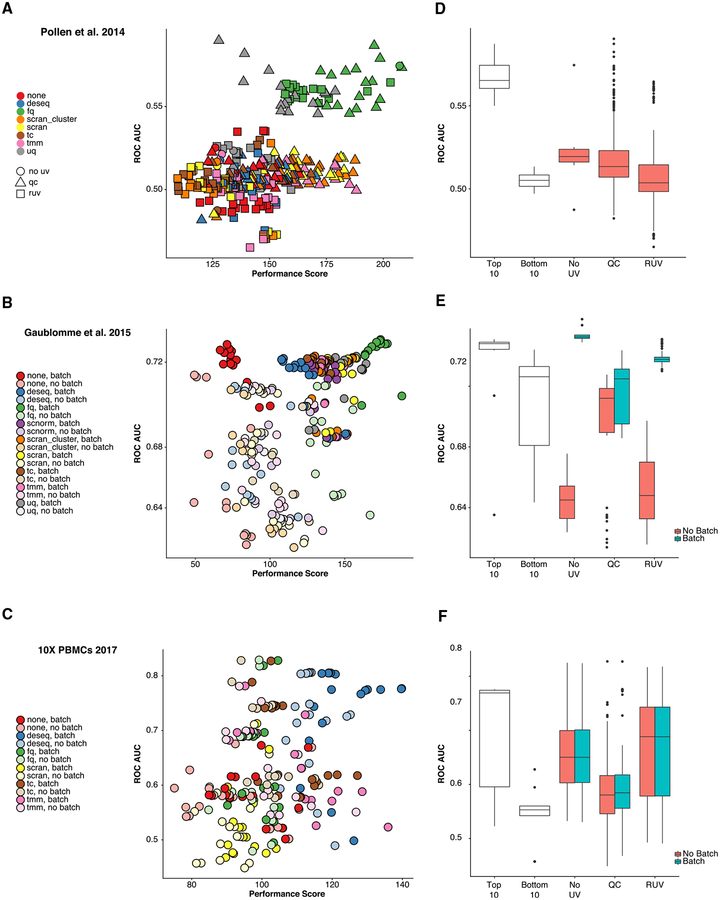
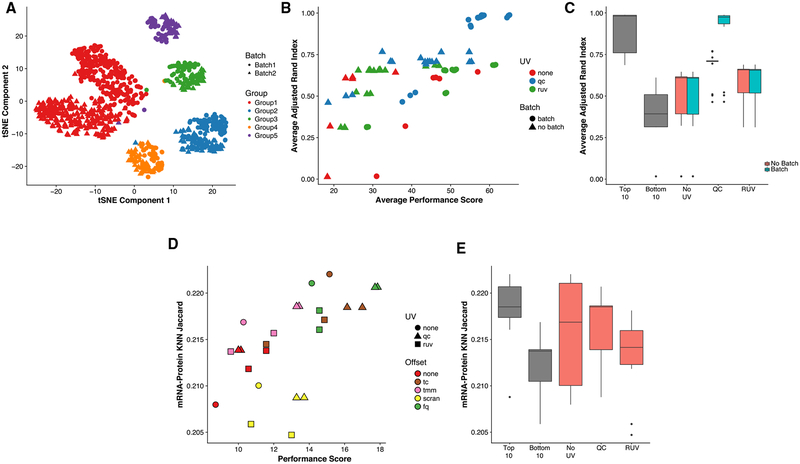
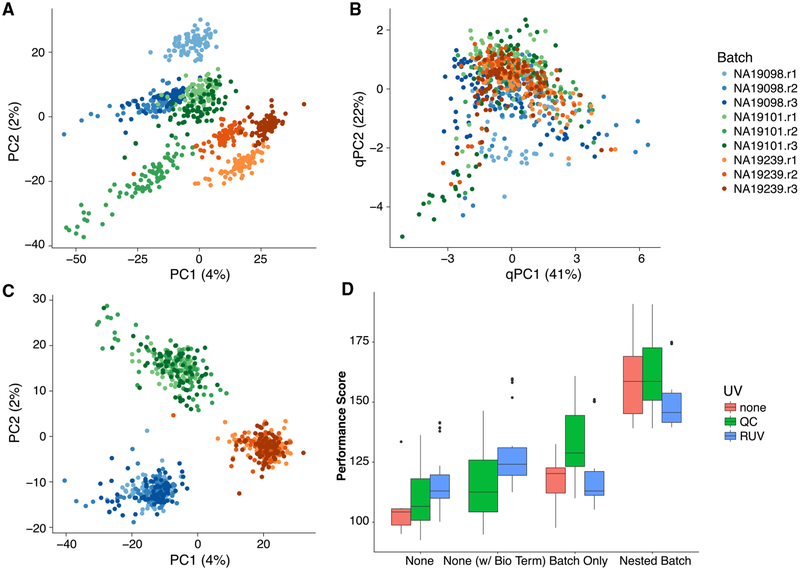
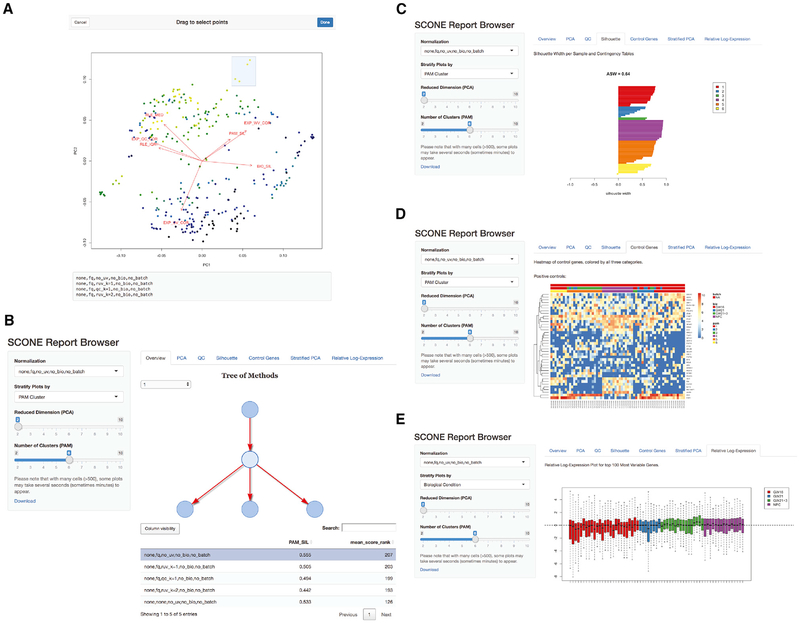
References
-
- Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Teichmann SA, Marioni JC, and Stegle O (2015). Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol 33, 155–160. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
