

An actionable sterol-regulated feedback loop modulates statin sensitivity in prostate cancer
- PMID: 31023626
- PMCID: PMC6600047
- DOI: 10.1016/j.molmet.2019.04.003
An actionable sterol-regulated feedback loop modulates statin sensitivity in prostate cancer
Abstract
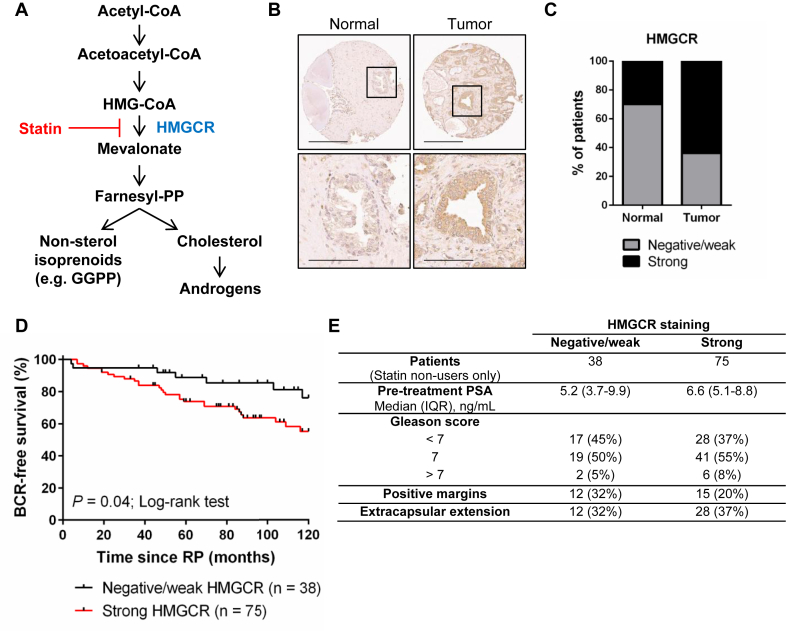
Objective: The statin family of cholesterol-lowering drugs has been shown to induce tumor-specific apoptosis by inhibiting the rate-limiting enzyme of the mevalonate (MVA) pathway, HMG-CoA reductase (HMGCR). Accumulating evidence suggests that statin use may delay prostate cancer (PCa) progression in a subset of patients; however, the determinants of statin drug sensitivity in PCa remain unclear. Our goal was to identify molecular features of statin-sensitive PCa and opportunities to potentiate statin-induced PCa cell death.
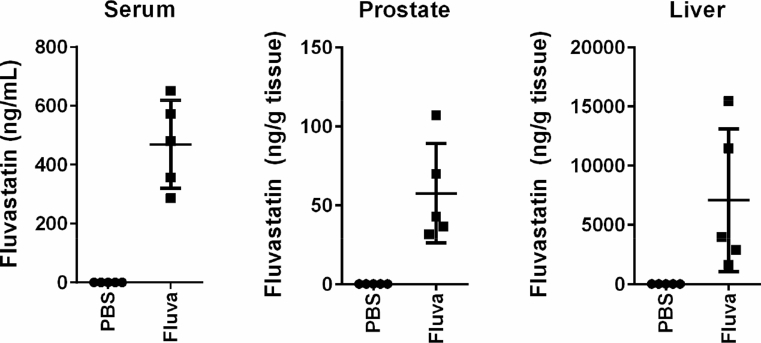
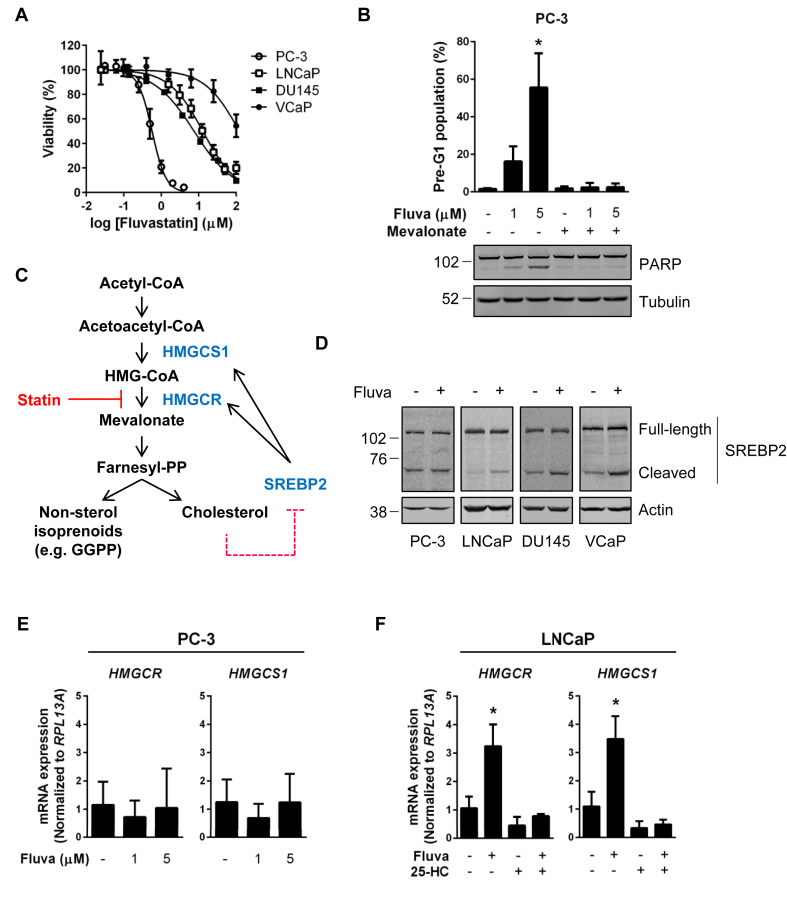
Methods: Deregulation of HMGCR expression in PCa was evaluated by immunohistochemistry. The response of PCa cell lines to fluvastatin-mediated HMGCR inhibition was assessed using cell viability and apoptosis assays. Activation of the sterol-regulated feedback loop of the MVA pathway, which was hypothesized to modulate statin sensitivity in PCa, was also evaluated. Inhibition of this statin-induced feedback loop was performed using RNA interference or small molecule inhibitors. The achievable levels of fluvastatin in mouse prostate tissue were measured using liquid chromatography-mass spectrometry.
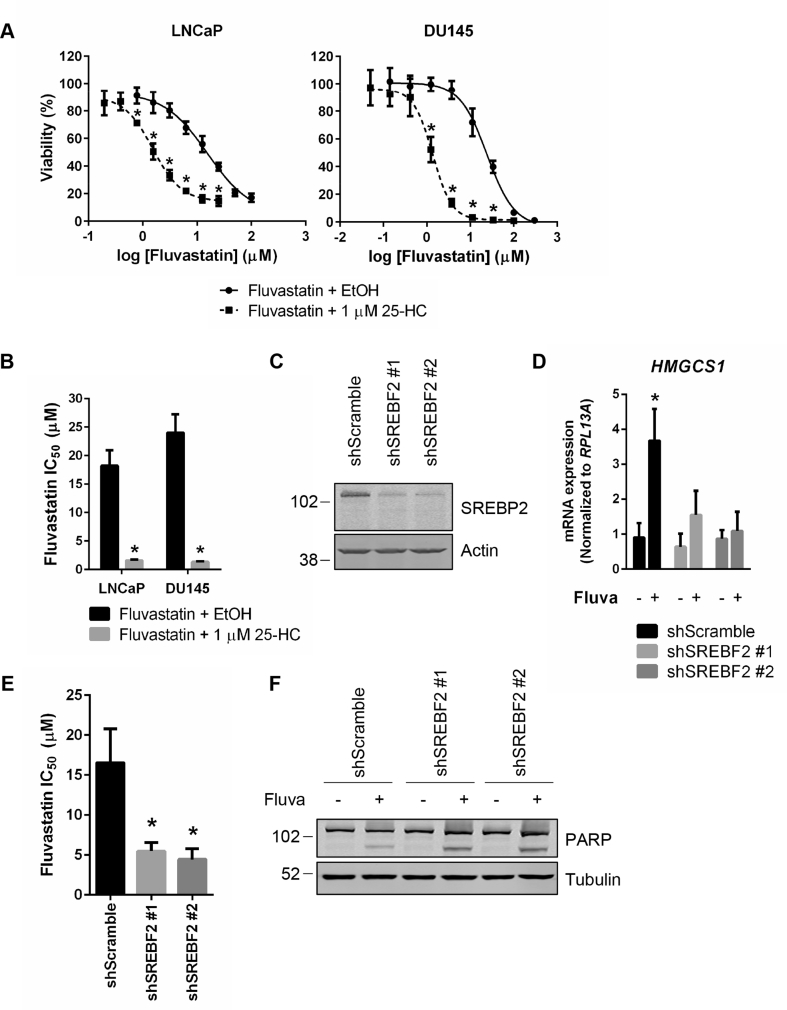
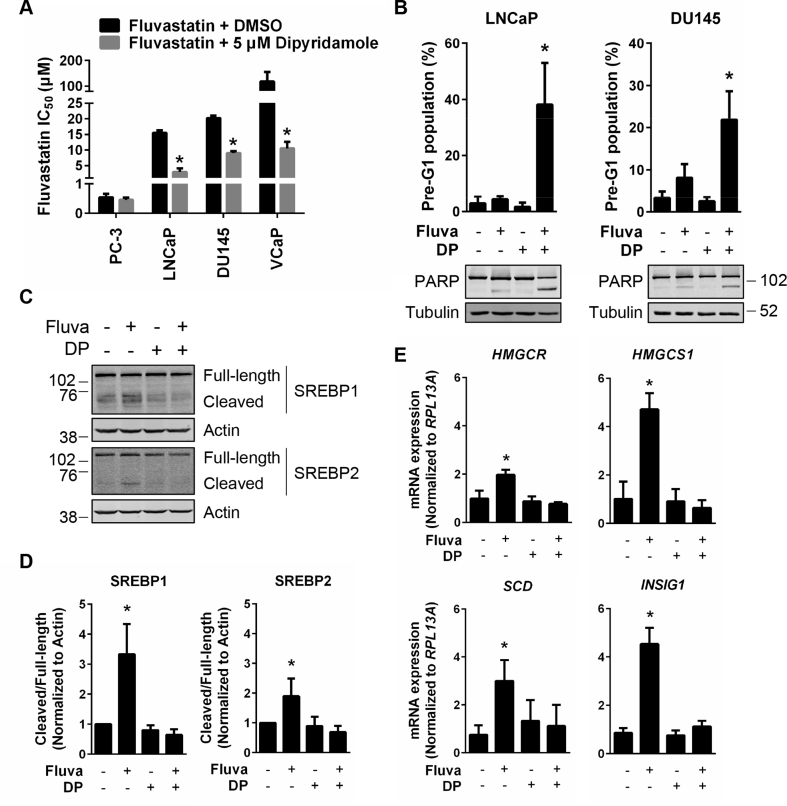
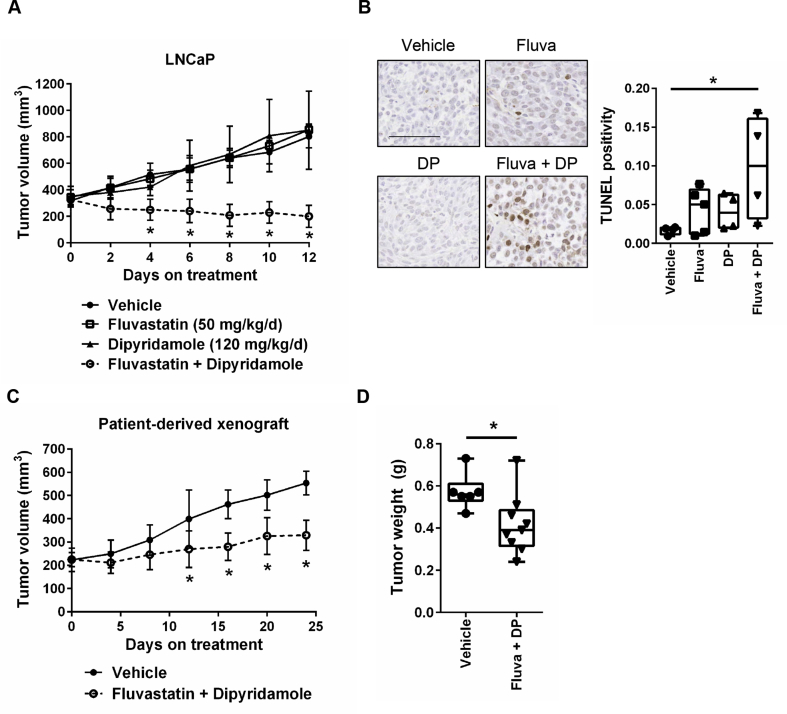
Results: High HMGCR expression in PCa was associated with poor prognosis; however, not all PCa cell lines underwent apoptosis in response to treatment with physiologically-achievable concentrations of fluvastatin. Rather, most cell lines initiated a feedback response mediated by sterol regulatory element-binding protein 2 (SREBP2), which led to the further upregulation of HMGCR and other lipid metabolism genes. Overcoming this feedback mechanism by knocking down or inhibiting SREBP2 potentiated fluvastatin-induced PCa cell death. Notably, we demonstrated that this feedback loop is pharmacologically-actionable, as the drug dipyridamole can be used to block fluvastatin-induced SREBP activation and augment apoptosis in statin-insensitive PCa cells.
Conclusion: Our study implicates statin-induced SREBP2 activation as a PCa vulnerability that can be exploited for therapeutic purposes using clinically-approved agents.
Keywords: Dipyridamole; Drug repurposing; Mevalonate pathway; Prostate cancer; Statins; Tumor metabolism.
Copyright © 2019 University Health Network. Published by Elsevier GmbH.. All rights reserved.
Figures
References
-
- Torre L.A., Bray F., Siegel R.L., Ferlay J., Lortet-Tieulent J., Jemal A. Global cancer statistics, 2012. CA. A Cancer Journal for Clinicians. 2015;65(2):87–108. - PubMed
-
- Sanda M.G., Dunn R.L., Michalski J., Sandler H.M., Northouse L., Hembroff L. Quality of life and satisfaction with outcome among prostate-cancer survivors. The New England Journal of Medicine. 2008;358(12):1250–1261. - PubMed
-
- Wong W.W.L., Dimitroulakos J., Minden M.D., Penn L.Z. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia. 2002;16(4):508–519. - PubMed
-
- Clendening J.W., Penn L.Z. Targeting tumor cell metabolism with statins. Oncogene. 2012;31(48):4967–4978. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
