

The impact of poly-A microsatellite heterologies in meiotic recombination
- PMID: 31023833
- PMCID: PMC6485458
- DOI: 10.26508/lsa.201900364
The impact of poly-A microsatellite heterologies in meiotic recombination
Abstract
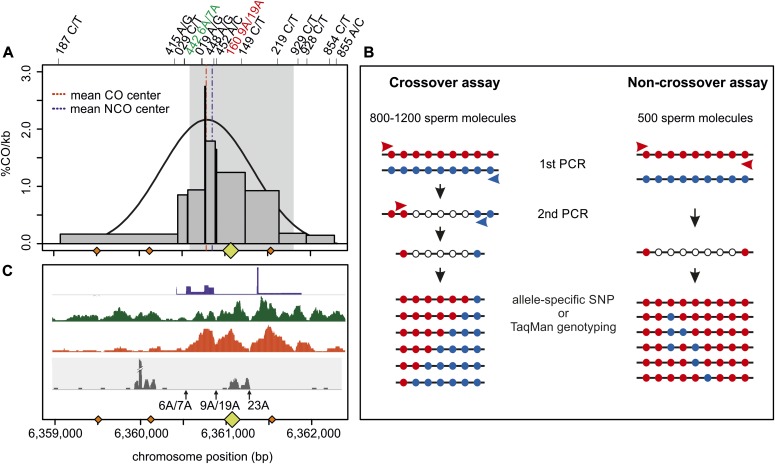
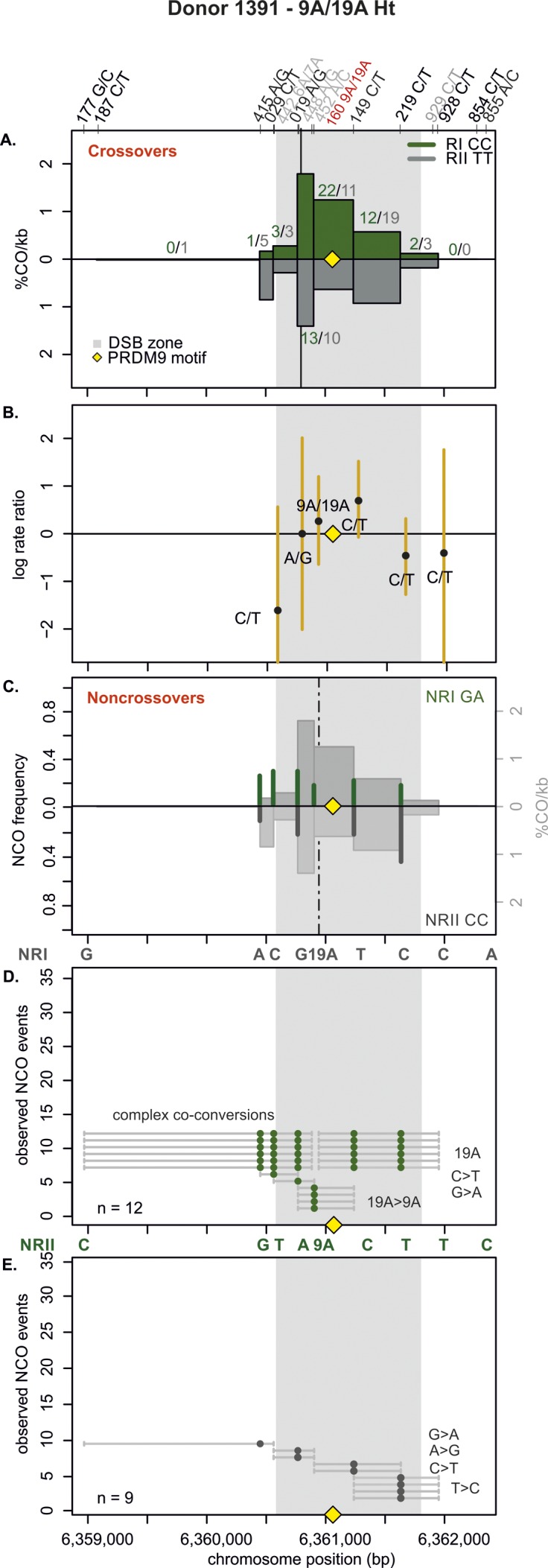
Meiotic recombination has strong, but poorly understood effects on short tandem repeat (STR) instability. Here, we screened thousands of single recombinant products with sperm typing to characterize the role of polymorphic poly-A repeats at a human recombination hotspot in terms of hotspot activity and STR evolution. We show that the length asymmetry between heterozygous poly-A's strongly influences the recombination outcome: a heterology of 10 A's (9A/19A) reduces the number of crossovers and elevates the frequency of non-crossovers, complex recombination products, and long conversion tracts. Moreover, the length of the heterology also influences the STR transmission during meiotic repair with a strong and significant insertion bias for the short heterology (6A/7A) and a deletion bias for the long heterology (9A/19A). In spite of this opposing insertion-/deletion-biased gene conversion, we find that poly-A's are enriched at human recombination hotspots that could have important consequences in hotspot activation.
© 2019 Heissl et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures




















References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources