

Adaptive Evolution within Gut Microbiomes of Healthy People
- PMID: 31028005
- PMCID: PMC6749991
- DOI: 10.1016/j.chom.2019.03.007
Adaptive Evolution within Gut Microbiomes of Healthy People
Abstract
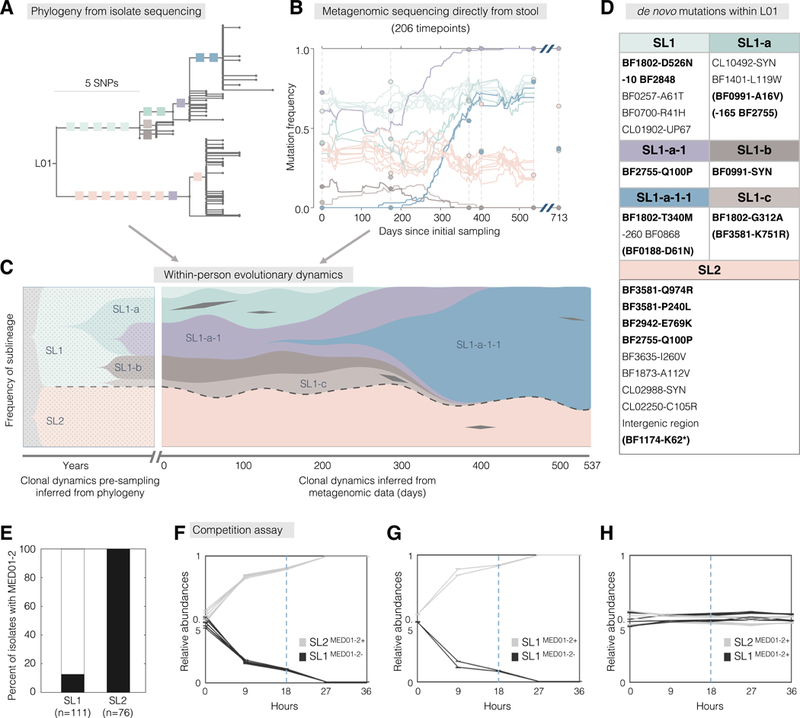
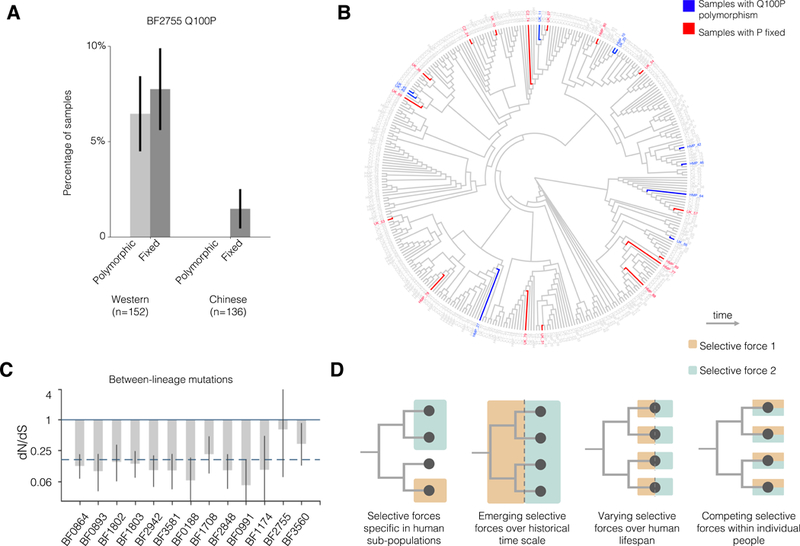
Natural selection shapes bacterial evolution in all environments. However, the extent to which commensal bacteria diversify and adapt within the human gut remains unclear. Here, we combine culture-based population genomics and metagenomics to investigate the within-microbiome evolution of Bacteroides fragilis. We find that intra-individual B. fragilis populations contain substantial de novo nucleotide and mobile element diversity, preserving years of within-person history. This history reveals multiple signatures of within-person adaptation, including parallel evolution in sixteen genes. Many of these genes are implicated in cell-envelope biosynthesis and polysaccharide utilization. Tracking evolutionary trajectories using near-daily metagenomic sampling, we find evidence for years-long coexistence in one subject despite adaptive dynamics. We used public metagenomes to investigate one adaptive mutation common in our cohort and found that it emerges frequently in Western, but not Chinese, microbiomes. Collectively, these results demonstrate that B. fragilis adapts within individual microbiomes, pointing to factors that promote long-term gut colonization.
Keywords: Bacteroides; adaptive evolution; de novo mutation; horizontal gene transfer; human microbiome; metagenomics; microbial evolution; microbial genomics; whole-genome sequencing.
Copyright © 2019 Elsevier Inc. All rights reserved.
Figures
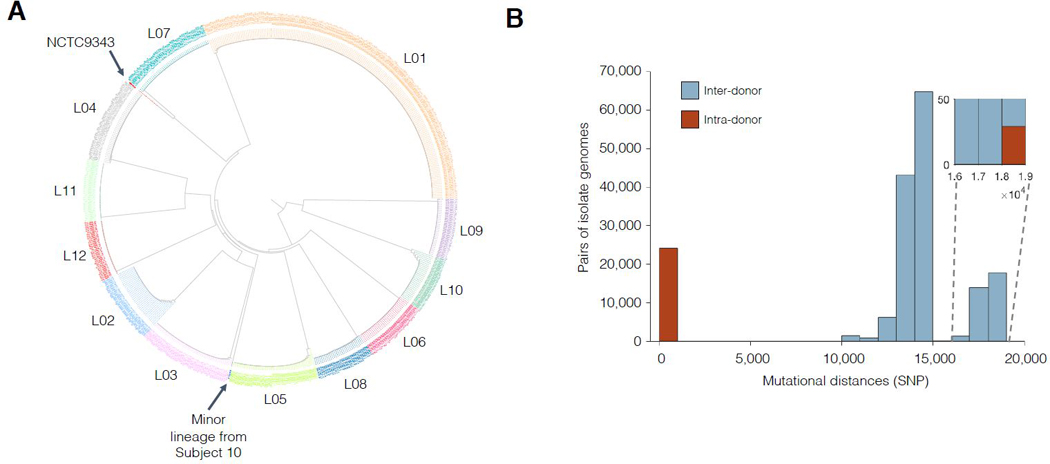
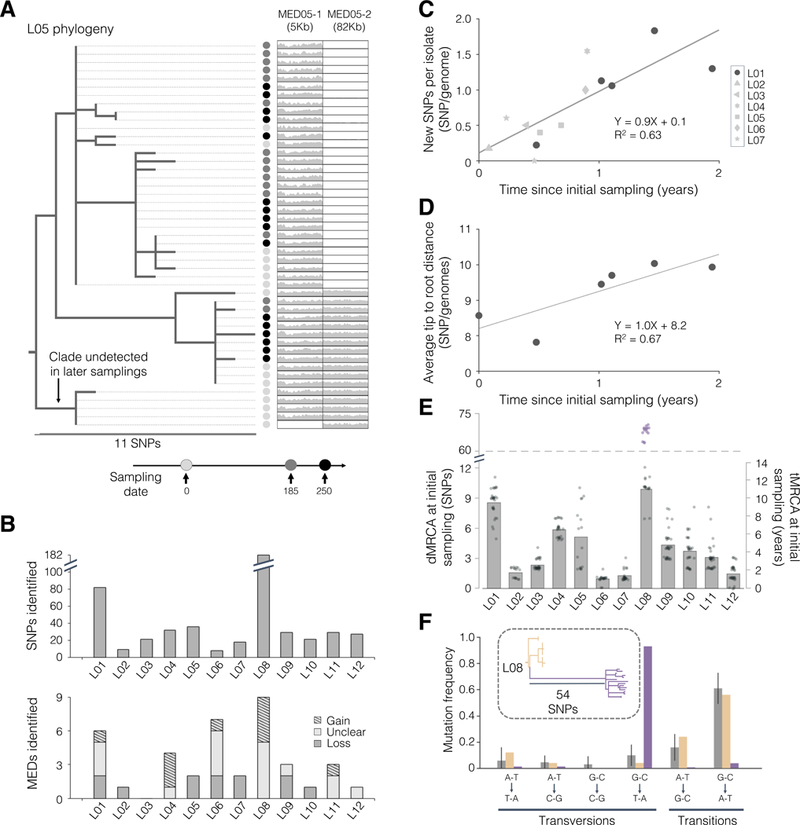
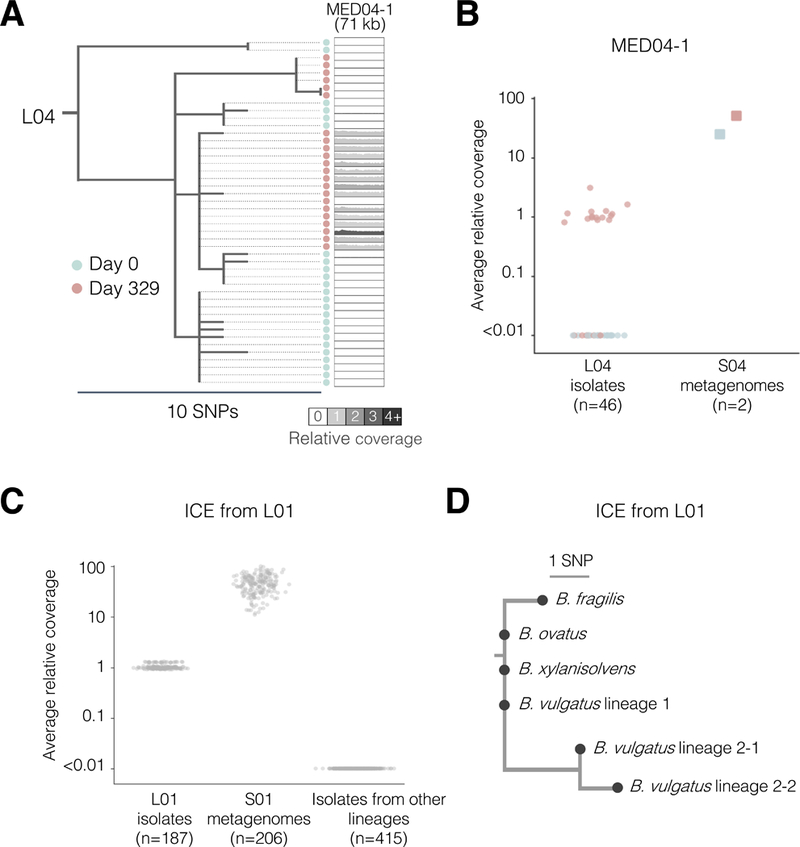
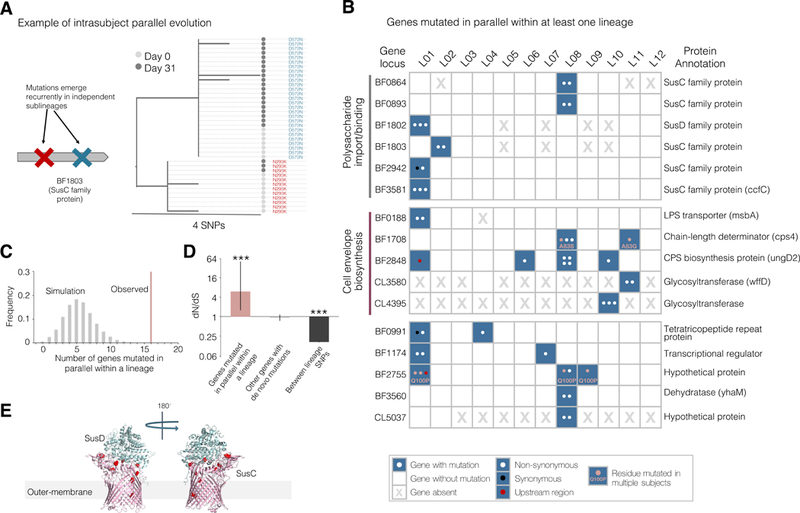
Comment in
-
Population dynamics of the human gut microbiome: change is the only constant.Genome Biol. 2019 Jul 31;20(1):150. doi: 10.1186/s13059-019-1775-3. Genome Biol. 2019. PMID: 31366367 Free PMC article.
References
-
- Bankevich Anton, Nurk Sergey, Antipov Dmitry, Gurevich Alexey A., Dvorkin Mikhail, Kulikov Alexander S., Lesin Valery M., et al. 2012. “SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing.” Journal of Computational Biology 19 (5): 455–77. 10.1089/cmb.2012.0021. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
