

Molecular mechanisms of arrhythmogenic cardiomyopathy
- PMID: 31028357
- PMCID: PMC6871180
- DOI: 10.1038/s41569-019-0200-7
Molecular mechanisms of arrhythmogenic cardiomyopathy
Abstract
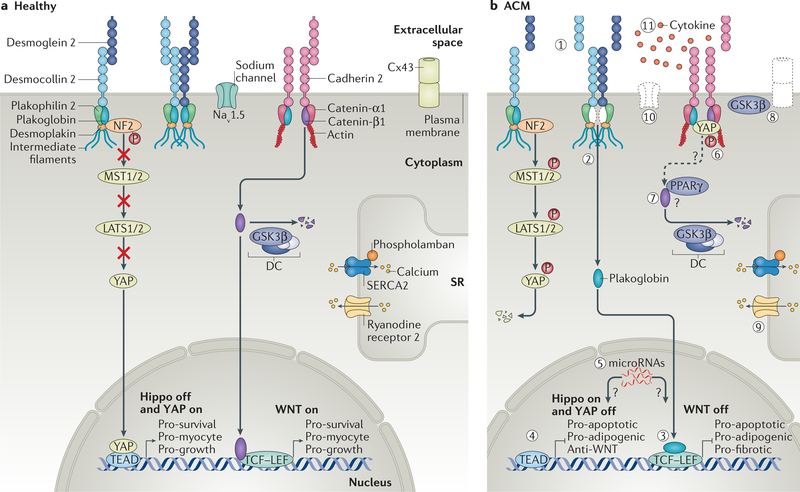
Arrhythmogenic cardiomyopathy is a genetic disorder characterized by the risk of life-threatening arrhythmias, myocardial dysfunction and fibrofatty replacement of myocardial tissue. Mutations in genes that encode components of desmosomes, the adhesive junctions that connect cardiomyocytes, are the predominant cause of arrhythmogenic cardiomyopathy and can be identified in about half of patients with the condition. However, the molecular mechanisms leading to myocardial destruction, remodelling and arrhythmic predisposition remain poorly understood. Through the development of animal, induced pluripotent stem cell and other models of disease, advances in our understanding of the pathogenic mechanisms of arrhythmogenic cardiomyopathy over the past decade have brought several signalling pathways into focus. These pathways include canonical and non-canonical WNT signalling, the Hippo-Yes-associated protein (YAP) pathway and transforming growth factor-β signalling. These studies have begun to identify potential therapeutic targets whose modulation has shown promise in preclinical models. In this Review, we summarize and discuss the reported molecular mechanisms underlying the pathogenesis of arrhythmogenic cardiomyopathy.
Figures


References
-
- Sen-Chowdhry S & McKenna WJ Reconciling the protean manifestations of arrhythmogenic cardiomyopathy. Circ. Arrhythm. Electrophysiol 3, 566–570 (2010). - PubMed
-
- Marcus FI et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation 65, 384–398 (1982). - PubMed
-
- Thiene G, Nava A, Corrado D, Rossi L & Pennelli N Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med 318, 129–133 (1988). - PubMed
-
- Sen-Chowdhry S, Morgan RD, Chambers JC & McKenna WJ Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu. Rev. Med 61, 233–253 (2010). - PubMed
-
- Peters S, Trummel M & Meyners W Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int. J. Cardiol 97, 499–501 (2004). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
