

The natural history of classic galactosemia: lessons from the GalNet registry
- PMID: 31029175
- PMCID: PMC6486996
- DOI: 10.1186/s13023-019-1047-z
The natural history of classic galactosemia: lessons from the GalNet registry
Abstract
Background: Classic galactosemia is a rare inborn error of carbohydrate metabolism, caused by a severe deficiency of the enzyme galactose-1-phosphate uridylyltransferase (GALT). A galactose-restricted diet has proven to be very effective to treat the neonatal life-threatening manifestations and has been the cornerstone of treatment for this severe disease. However, burdensome complications occur despite a lifelong diet. For rare diseases, a patient disease specific registry is fundamental to monitor the lifespan pathology and to evaluate the safety and efficacy of potential therapies. In 2014, the international Galactosemias Network (GalNet) developed a web-based patient registry for this disease, the GalNet Registry. The aim was to delineate the natural history of classic galactosemia based on a large dataset of patients.
Methods: Observational data derived from 15 countries and 32 centers including 509 patients were acquired between December 2014 and July 2018.
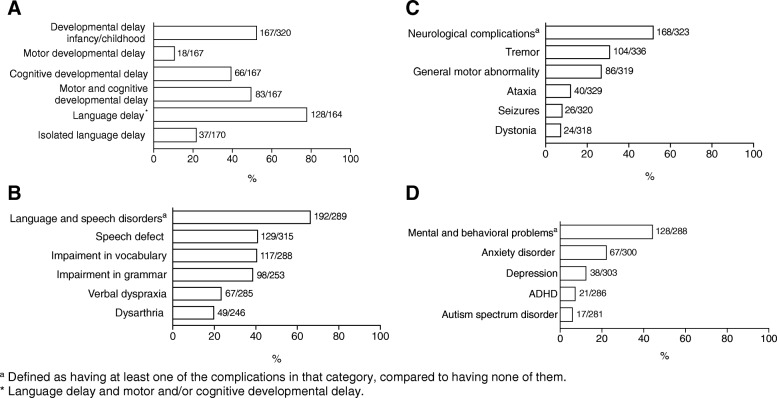
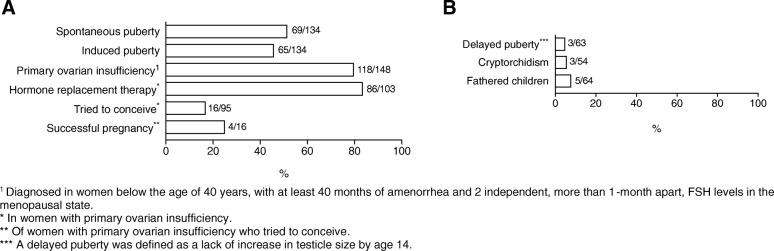
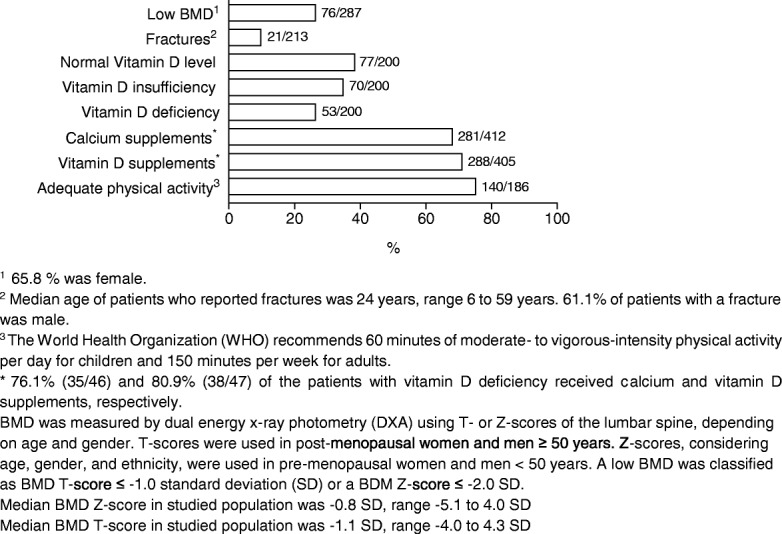
Results: Most affected patients experienced neonatal manifestations (79.8%) and despite following a diet developed brain impairments (85.0%), primary ovarian insufficiency (79.7%) and a diminished bone mineral density (26.5%). Newborn screening, age at onset of dietary treatment, strictness of the galactose-restricted diet, p.Gln188Arg mutation and GALT enzyme activity influenced the clinical picture. Detection by newborn screening and commencement of diet in the first week of life were associated with a more favorable outcome. A homozygous p.Gln188Arg mutation, GALT enzyme activity of ≤ 1% and strict galactose restriction were associated with a less favorable outcome.
Conclusion: This study describes the natural history of classic galactosemia based on the hitherto largest data set.
Keywords: GALT deficiency; Galactosemia; Galactosemia network; Natural history; Registry.
Conflict of interest statement
Ethics approval and consent to participate
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with Principles of the Declaration of Helsinki. The study was approved by the local ethics committee of the coordinating center, the Medical Ethical Committee (
Consent for publication
Consent was obtained from all patients for publication.
Competing interests
M.E.R-G., M.H., B.B., A.I.C., D.C, M.L.C., C.D., D.D., T.D., F.E., M.T.F., S.G., J.H., M.H., A.H., H.H.H., P.J., J.K., I.K., P.L., Y.E.L., J.G.L., D.M., D.M-W., K.Õ., D.R., I.A.R., S.S-B., K.M.S., A.T., C.T., R.Va., G.V., R.Vo., M.V., S.E.W., M.W-K., S.B.W., M.G., E.P.T., G.T.B. declare that they have no competing interests. A.M.B. has received a speakers fee and has been a member of advisory boards for Nutricia and Biomarin. E.M. has received travel funding, research grants and support from Nutricia UK.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Berry GT. GeneReviews®. 2000. Classic Galactosemia and Clinical Variant Galactosemia. - PubMed
-
- Coss KP, Doran PP, Owoeye C, Codd MB, Hamid N, Mayne PD, et al. Classical Galactosaemia in Ireland: incidence, complications and outcomes of treatment. J Inherit Metab Dis. 2013;36(1):21. - PubMed
-
- Calderon FR, Phansalkar AR, Crockett DK, Miller M, Mao R. Mutation database for the galactose-1-phosphate uridyltransferase (GALT) gene. Hum Mutat. 2007;28(10):939. - PubMed
-
- Mason HH, Turner ME. Chronic galactemia, report of case with studies on carbohydrates. Am J Dis Child. 1935;50(2):359.
-
- Isselbacher KJ, Anderson EP, Kurahashi K, Kalckar HM. Congenital galactosemia, a single enzymatic block in galactose metabolism. Science. 1956;123(3198):635–636. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
