

Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities
- PMID: 31031003
- PMCID: PMC6768830
- DOI: 10.1016/j.cell.2019.03.030
Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities
Abstract
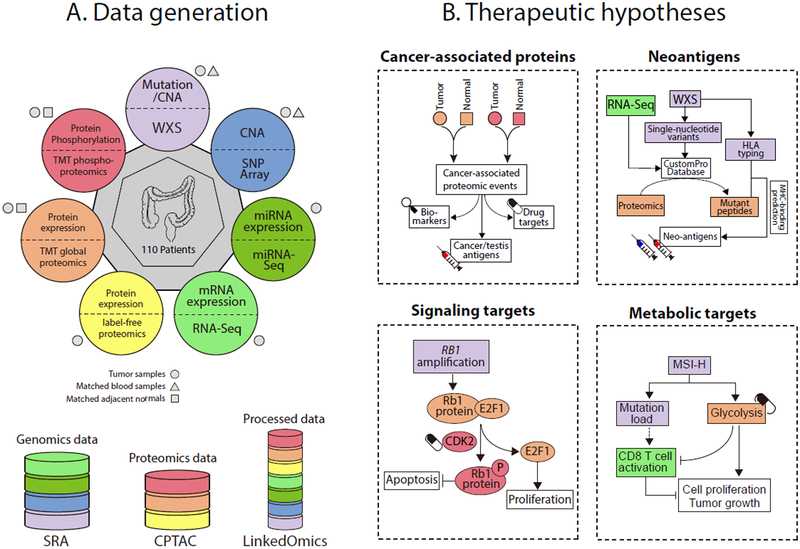
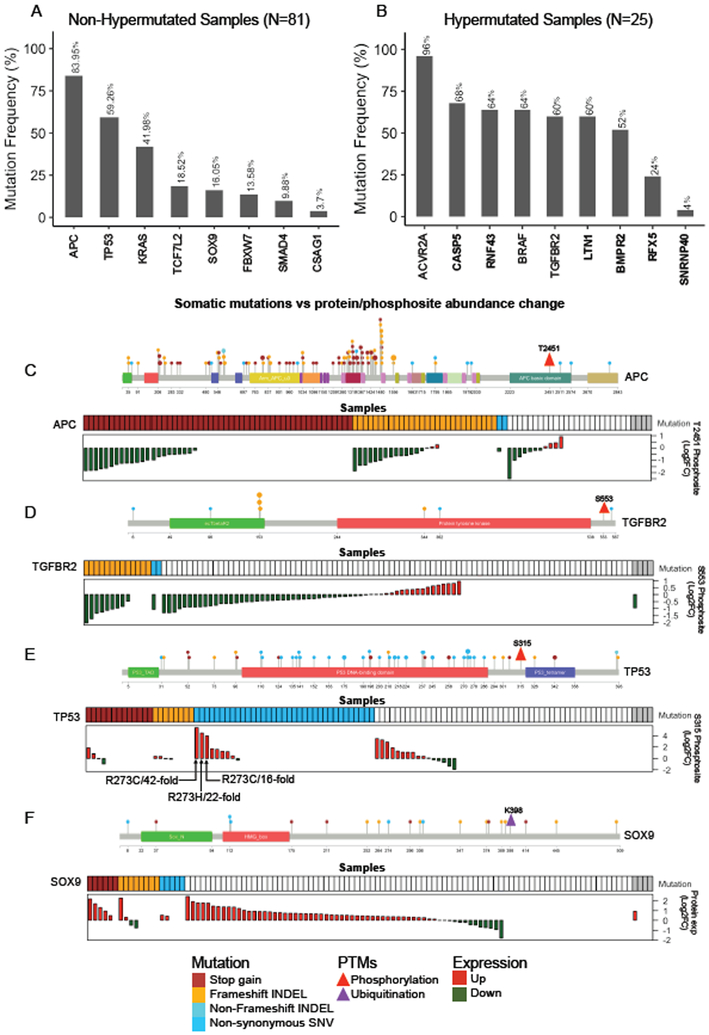
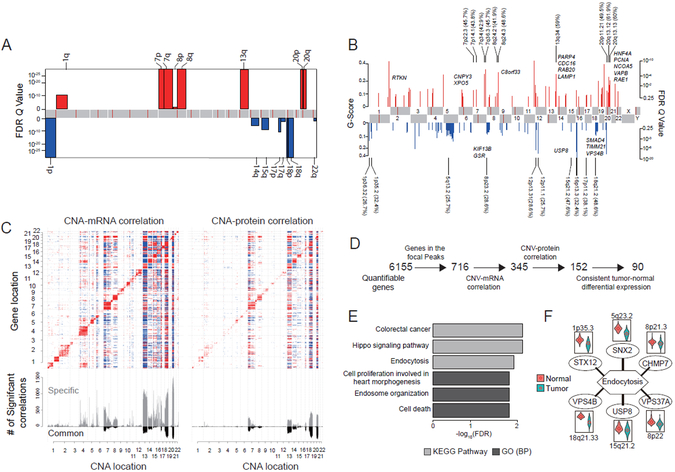
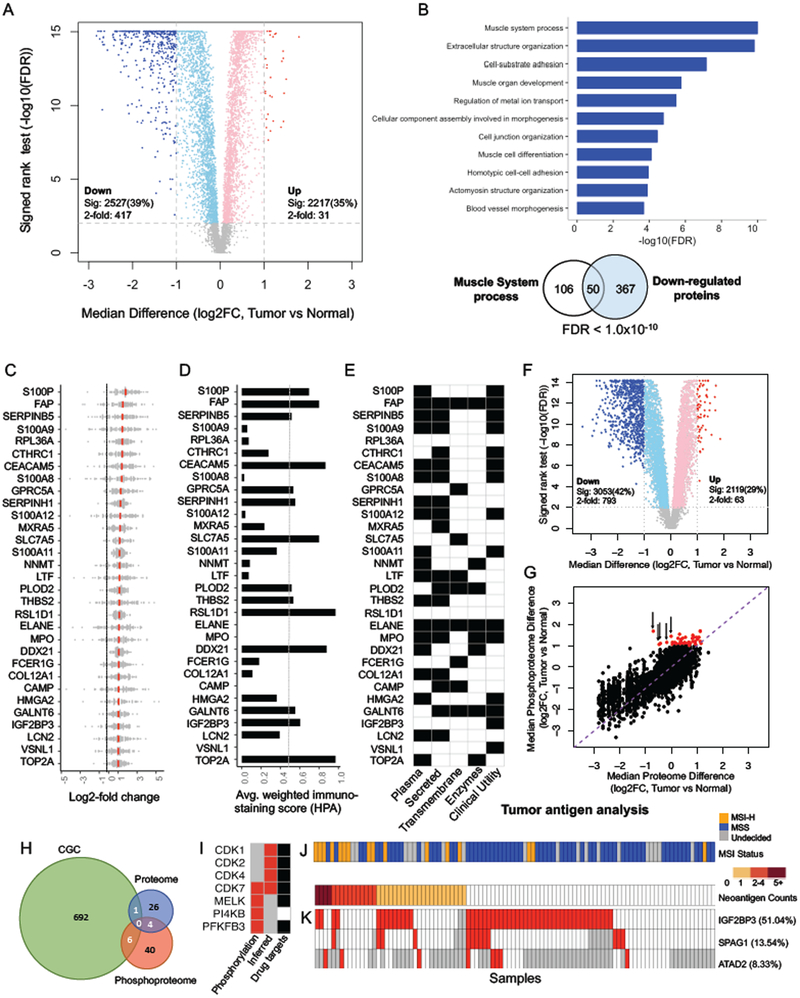
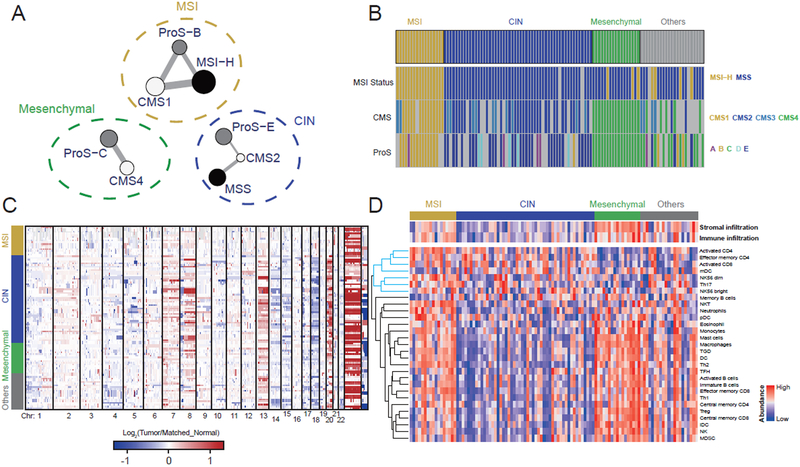
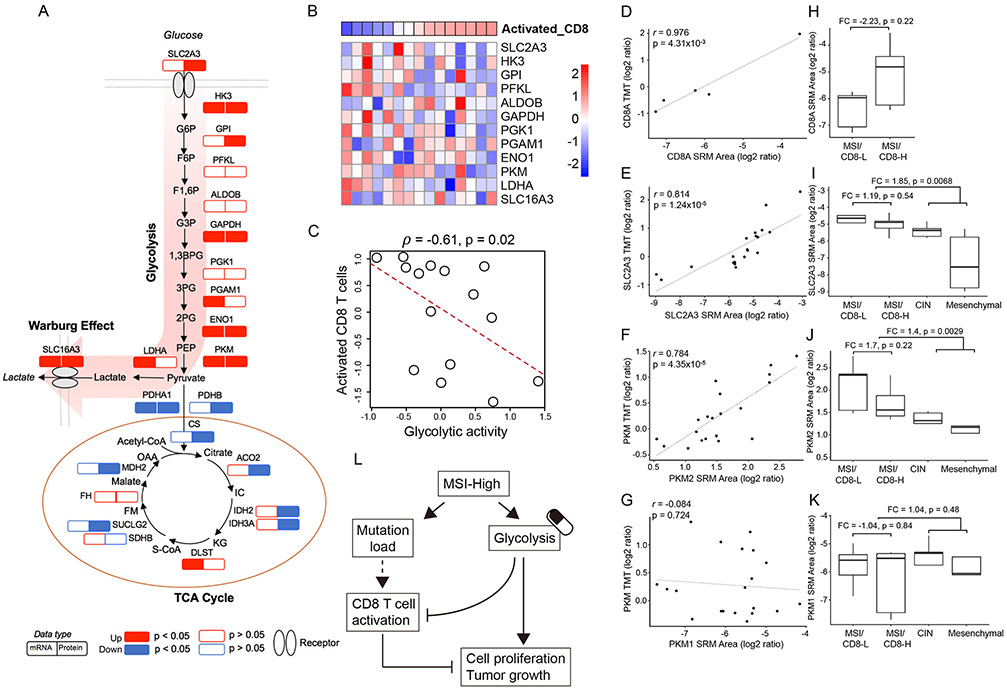
We performed the first proteogenomic study on a prospectively collected colon cancer cohort. Comparative proteomic and phosphoproteomic analysis of paired tumor and normal adjacent tissues produced a catalog of colon cancer-associated proteins and phosphosites, including known and putative new biomarkers, drug targets, and cancer/testis antigens. Proteogenomic integration not only prioritized genomically inferred targets, such as copy-number drivers and mutation-derived neoantigens, but also yielded novel findings. Phosphoproteomics data associated Rb phosphorylation with increased proliferation and decreased apoptosis in colon cancer, which explains why this classical tumor suppressor is amplified in colon tumors and suggests a rationale for targeting Rb phosphorylation in colon cancer. Proteomics identified an association between decreased CD8 T cell infiltration and increased glycolysis in microsatellite instability-high (MSI-H) tumors, suggesting glycolysis as a potential target to overcome the resistance of MSI-H tumors to immune checkpoint blockade. Proteogenomics presents new avenues for biological discoveries and therapeutic development.
Keywords: RB1; SOX9; biomarkers; colon cancer; drug targets; glycolysis; immune evasion; proteogenomics; proteomics; tumor antigen.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests
The authors declare no competing interests.
Figures

Comment in
-
Atlas Drugged.Cell. 2019 May 2;177(4):803-805. doi: 10.1016/j.cell.2019.04.023. Cell. 2019. PMID: 31051104
References
-
- Akiyama H, Kamitani T, Yang X, Kandyil R, Bridgewater LC, Fellous M, Mori-Akiyama Y, and de Crombrugghe B (2005). The transcription factor Sox9 is degraded by the ubiquitin-proteasome system and stabilized by a mutation in a ubiquitin-target site. Matrix Biol 23, 499–505. - PubMed
-
- Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, Waldner MJ, Bindea G, Mlecnik B, Galon J, et al. (2015). Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol 16, 64. - PMC - PubMed
-
- Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, and Bray F (2017). Global patterns and trends in colorectal cancer incidence and mortality. Gut 66, 683–691. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
