

Microbiota of the Gut-Lymph Node Axis: Depletion of Mucosa-Associated Segmented Filamentous Bacteria and Enrichment of Methanobrevibacter by Colistin Sulfate and Linco-Spectin in Pigs
- PMID: 31031713
- PMCID: PMC6470194
- DOI: 10.3389/fmicb.2019.00599
Microbiota of the Gut-Lymph Node Axis: Depletion of Mucosa-Associated Segmented Filamentous Bacteria and Enrichment of Methanobrevibacter by Colistin Sulfate and Linco-Spectin in Pigs
Erratum in
-
Erratum: Microbiota of the Gut-Lymph Node Axis: Depletion of Mucosa-Associated Segmented Filamentous Bacteria and Enrichment of Methanobrevibacter by Colistin Sulfate and Linco-Spectin in Pigs.Front Microbiol. 2020 May 26;11:1051. doi: 10.3389/fmicb.2020.01051. eCollection 2020. Front Microbiol. 2020. PMID: 32528449 Free PMC article.
Abstract
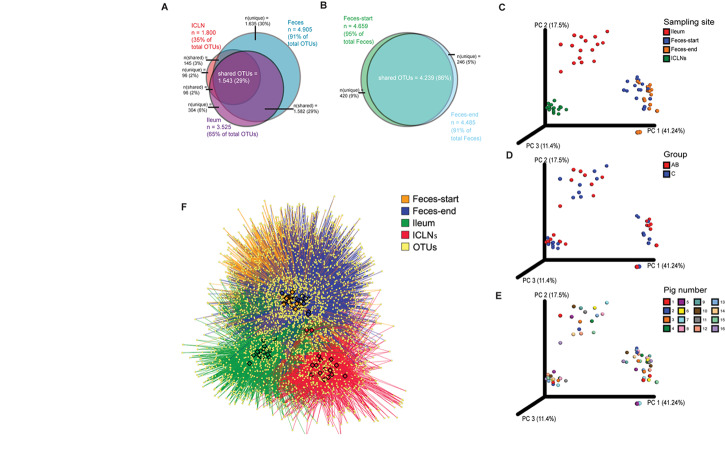
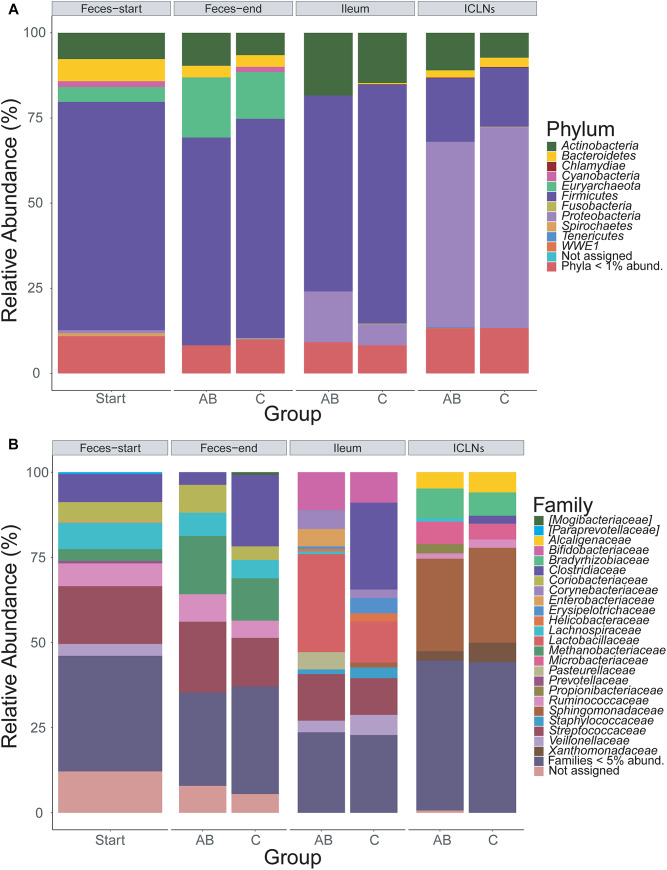
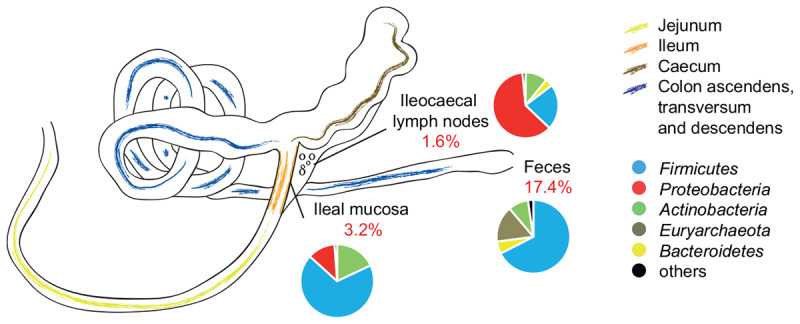
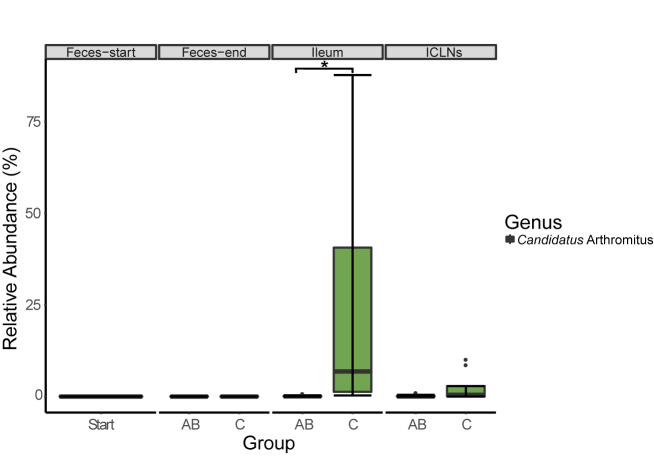
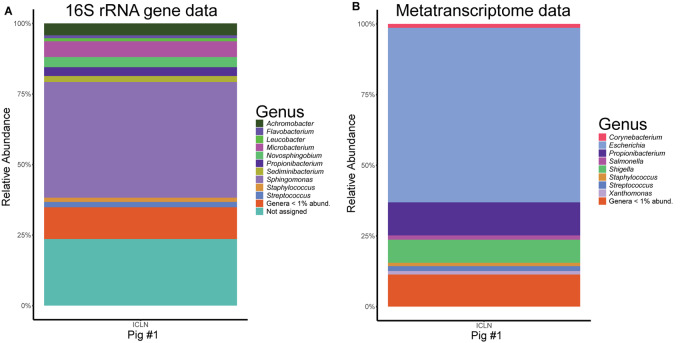
Microorganisms are translocated from the gut to lymphatic tissues via immune cells, thereby challenging and training the mammalian immune system. Antibiotics alter the gut microbiome and consecutively might also affect the corresponding translocation processes, resulting in an imbalanced state between the intestinal microbiota and the host. Hence, understanding the variant effects of antibiotics on the microbiome of gut-associated tissues is of vital importance for maintaining metabolic homeostasis and animal health. In the present study, we analyzed the microbiome of (i) pig feces, ileum, and ileocecal lymph nodes under the influence of antibiotics (Linco-Spectin and Colistin sulfate) using 16S rRNA gene sequencing for high-resolution community profiling and (ii) ileocecal lymph nodes in more detail with two additional methodological approaches, i.e., cultivation of ileocecal lymph node samples and (iii) metatranscriptome sequencing of a single lymph node sample. Supplementation of medicated feed showed a local effect on feces and ileal mucosa-associated microbiomes. Pigs that received antibiotics harbored significantly reduced amounts of segmented filamentous bacteria (SFB) along the ileal mucosa (p = 0.048; 199.17-fold change) and increased amounts of Methanobrevibacter, a methanogenic Euryarchaeote in fecal samples (p = 0.005; 20.17-fold change) compared to the control group. Analysis of the porcine ileocecal lymph node microbiome exposed large differences between the viable and the dead fraction of microorganisms and the microbiome was altered to a lesser extent by antibiotics compared with feces and ileum. The core microbiome of lymph nodes was constituted mainly of Proteobacteria. RNA-sequencing of a single lymph node sample unveiled transcripts responsible for amino acid and carbohydrate metabolism as well as protein turnover, DNA replication and signal transduction. The study presented here is the first comparative study of microbial communities in feces, ileum, and its associated ileocecal lymph nodes. In each analyzed site, we identified specific phylotypes susceptible to antibiotic treatment that can have profound impacts on the host physiological and immunological state, or even on global biogeochemical cycles. Our results indicate that pathogenic bacteria, e.g., enteropathogenic Escherichia coli, could escape antibiotic treatment by translocating to lymph nodes. In general ileocecal lymph nodes harbor a more diverse and active community of microorganisms than previously assumed.
Keywords: 16S rRNA gene; antibiotics; gut microbiota; ileum; lymph nodes; metatranscriptome; microbiome; segmented filamentous bacteria.
Figures
Similar articles
-
Alterations of the Viable Ileal Microbiota of the Gut Mucosa-Lymph Node Axis in Pigs Fed Phytase and Lactic Acid-Treated Cereals.Appl Environ Microbiol. 2020 Feb 3;86(4):e02128-19. doi: 10.1128/AEM.02128-19. Print 2020 Feb 3. Appl Environ Microbiol. 2020. PMID: 31757823 Free PMC article.
-
Erratum: Microbiota of the Gut-Lymph Node Axis: Depletion of Mucosa-Associated Segmented Filamentous Bacteria and Enrichment of Methanobrevibacter by Colistin Sulfate and Linco-Spectin in Pigs.Front Microbiol. 2020 May 26;11:1051. doi: 10.3389/fmicb.2020.01051. eCollection 2020. Front Microbiol. 2020. PMID: 32528449 Free PMC article.
-
Changes in the Ileal, but Not Fecal, Microbiome in Response to Increased Dietary Protein Level and Enterotoxigenic Escherichia coli Exposure in Pigs.Appl Environ Microbiol. 2019 Sep 17;85(19):e01252-19. doi: 10.1128/AEM.01252-19. Print 2019 Oct 1. Appl Environ Microbiol. 2019. PMID: 31324635 Free PMC article.
-
Segmented Filamentous Bacteria - Metabolism Meets Immunity.Front Microbiol. 2018 Aug 24;9:1991. doi: 10.3389/fmicb.2018.01991. eCollection 2018. Front Microbiol. 2018. PMID: 30197636 Free PMC article. Review.
-
Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances.Front Microbiol. 2016 Jan 12;6:1543. doi: 10.3389/fmicb.2015.01543. eCollection 2015. Front Microbiol. 2016. PMID: 26793178 Free PMC article. Review.
Cited by
-
Relationships among Fecal, Air, Oral, and Tracheal Microbial Communities in Pigs in a Respiratory Infection Disease Model.Microorganisms. 2021 Jan 27;9(2):252. doi: 10.3390/microorganisms9020252. Microorganisms. 2021. PMID: 33513772 Free PMC article.
-
Bacteriological, cytological, and molecular investigation of Corynebacterium pseudotuberculosis, mycobacteria, and other bacteria in caseous lymphadenitis and healthy lymph nodes of slaughtered sheep.Braz J Microbiol. 2021 Mar;52(1):431-438. doi: 10.1007/s42770-020-00403-0. Epub 2020 Nov 13. Braz J Microbiol. 2021. PMID: 33185852 Free PMC article.
-
Maximum levels of cross-contamination for 24 antimicrobial active substances in non-target feed. Part 9: Polymyxins: colistin.EFSA J. 2021 Oct 26;19(10):e06861. doi: 10.2903/j.efsa.2021.6861. eCollection 2021 Oct. EFSA J. 2021. PMID: 34729089 Free PMC article.
-
Maximum levels of cross-contamination for 24 antimicrobial active substances in non-target feed. Part 2: Aminoglycosides/aminocyclitols: apramycin, paromomycin, neomycin and spectinomycin.EFSA J. 2021 Oct 26;19(10):e06853. doi: 10.2903/j.efsa.2021.6853. eCollection 2021 Oct. EFSA J. 2021. PMID: 34729082 Free PMC article.
-
Alterations of the Viable Ileal Microbiota of the Gut Mucosa-Lymph Node Axis in Pigs Fed Phytase and Lactic Acid-Treated Cereals.Appl Environ Microbiol. 2020 Feb 3;86(4):e02128-19. doi: 10.1128/AEM.02128-19. Print 2020 Feb 3. Appl Environ Microbiol. 2020. PMID: 31757823 Free PMC article.
References
-
- Bernard K. A., Wiebe D., Burdz T., Reimer A., Ng B., Singh C., et al. (2010). Assignment of Brevibacterium stationis (ZoBell and Upham 1944) Breed 1953 to the genus Corynebacterium, as Corynebacterium stationis comb. nov., and emended description of the genus Corynebacterium to include isolates that can alkalinize citrate. Int. J. Syst. Evol. Microbiol. 60 874–879. 10.1099/ijs.0.012641-0 - DOI - PubMed
LinkOut - more resources
Full Text Sources