

Experimental Models of Brugada syndrome
- PMID: 31032819
- PMCID: PMC6539778
- DOI: 10.3390/ijms20092123
Experimental Models of Brugada syndrome
Abstract
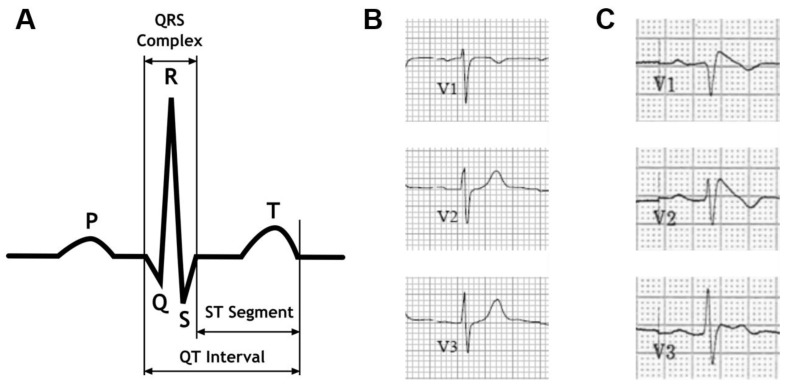
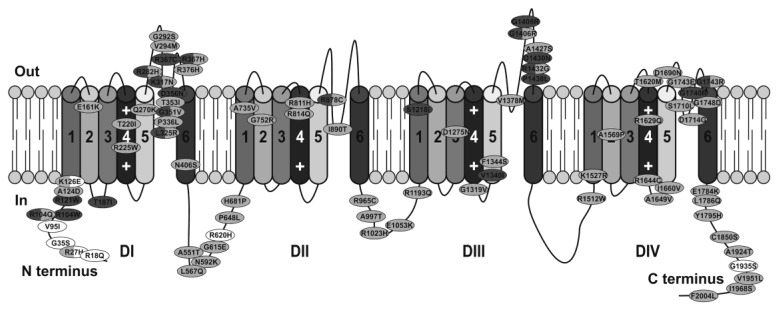
Brugada syndrome is an inherited, rare cardiac arrhythmogenic disease, associated with sudden cardiac death. It accounts for up to 20% of sudden deaths in patients without structural cardiac abnormalities. The majority of mutations involve the cardiac sodium channel gene SCN5A and give rise to classical abnormal electrocardiogram with ST segment elevation in the right precordial leads V1 to V3 and a predisposition to ventricular fibrillation. The pathophysiological mechanisms of Brugada syndrome have been investigated using model systems including transgenic mice, canine heart preparations, and expression systems to study different SCN5A mutations. These models have a number of limitations. The recent development of pluripotent stem cell technology creates an opportunity to study cardiomyocytes derived from patients and healthy individuals. To date, only a few studies have been done using Brugada syndrome patient-specific iPS-CM, which have provided novel insights into the mechanisms and pathophysiology of Brugada syndrome. This review provides an evaluation of the strengths and limitations of each of these model systems and summarizes the key mechanisms that have been identified to date.
Keywords: Brugada syndrome; SCN5A; induced pluripotent stem cells (iPS); model systems.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Coronel R., Casini S., Koopmann T.T., Wilms-Schopman F.J., Verkerk A.O., de Groot J.R., Bhuiyan Z., Bezzina C.R., Veldkamp M.W., Linnenbank A.C., et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation. 2005;112:2769–2777. doi: 10.1161/CIRCULATIONAHA.105.532614. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
