

BEAGLE 3: Improved Performance, Scaling, and Usability for a High-Performance Computing Library for Statistical Phylogenetics
- PMID: 31034053
- PMCID: PMC6802572
- DOI: 10.1093/sysbio/syz020
BEAGLE 3: Improved Performance, Scaling, and Usability for a High-Performance Computing Library for Statistical Phylogenetics
Abstract
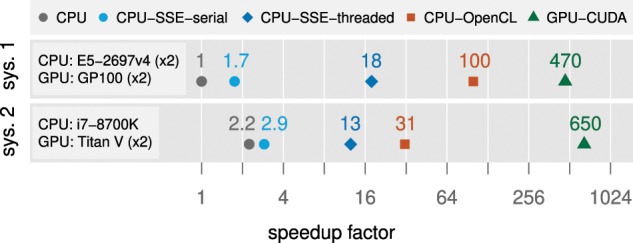
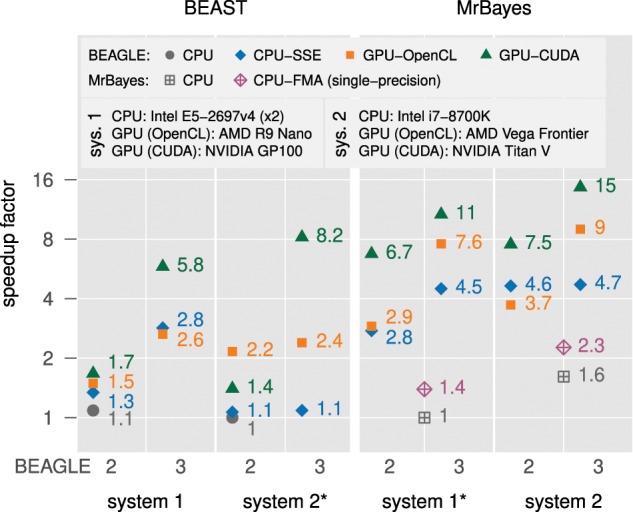
BEAGLE is a high-performance likelihood-calculation library for phylogenetic inference. The BEAGLE library defines a simple, but flexible, application programming interface (API), and includes a collection of efficient implementations for calculation under a variety of evolutionary models on different hardware devices. The library has been integrated into recent versions of popular phylogenetics software packages including BEAST and MrBayes and has been widely used across a diverse range of evolutionary studies. Here, we present BEAGLE 3 with new parallel implementations, increased performance for challenging data sets, improved scalability, and better usability. We have added new OpenCL and central processing unit-threaded implementations to the library, allowing the effective utilization of a wider range of modern hardware. Further, we have extended the API and library to support concurrent computation of independent partial likelihood arrays, for increased performance of nucleotide-model analyses with greater flexibility of data partitioning. For better scalability and usability, we have improved how phylogenetic software packages use BEAGLE in multi-GPU (graphics processing unit) and cluster environments, and introduced an automated method to select the fastest device given the data set, evolutionary model, and hardware. For application developers who wish to integrate the library, we also have developed an online tutorial. To evaluate the effect of the improvements, we ran a variety of benchmarks on state-of-the-art hardware. For a partitioned exemplar analysis, we observe run-time performance improvements as high as 5.9-fold over our previous GPU implementation. BEAGLE 3 is free, open-source software licensed under the Lesser GPL and available at https://beagle-dev.github.io.
Keywords: Bayesian phylogenetics; GPU; maximum likelihood; multicore processing; parallel computing.
© The Author(s) 2019. Published by Oxford University Press, on behalf of the Society of Systematic Biologists.
Figures



References
-
- Altekar G., Dwarkadas S., Huelsenbeck J.P., Ronquist F.. 2004. Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics. 20:407–415. - PubMed
-
- Ayres D.L., Cummings M.P.. 2017a. Configuring concurrent computation of phylogenetic partial likelihoods: accelerating analyses using the BEAGLE library In: Ibrahim S., Choo K., Yan Z., Pedrycz W., editors. Algorithms and architectures for parallel processing. Vol. 10393 of Lect. Notes Comput. Sc. Helsinki, Finland: ICA3PP; 2017. p. 533–547.
-
- Ayres D.L., Cummings M.P.. 2017b. Heterogeneous hardware support in BEAGLE, a high-performance computing library for statistical phylogenetics. 2017 46th International Conference on Parallel Processing Workshops (ICPPW), Bristol, 2017 p. 23–32.
-
- Ayres D.L., Cummings M.P.. 2018. Rerooting trees increases opportunities for concurrent computation and results in markedly improved performance for phylogenetic inference. 2018 IEEE International Parallel and Distributed Processing Symposium Workshops (IPDPSW). Vancouver, BC, 2018. p. 247–256.
-
- Ayres D.L., Darling A., Zwickl D.J., Beerli P., Holder M.T., Lewis P.O., Huelsenbeck J.P., Ronquist F., Swofford D.L., Cummings M.P., Rambaut A., Suchard M.A.. 2012. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 61:170–173. - PMC - PubMed
