

Measuring sequencer size bias using REcount: a novel method for highly accurate Illumina sequencing-based quantification
- PMID: 31036053
- PMCID: PMC6489363
- DOI: 10.1186/s13059-019-1691-6
Measuring sequencer size bias using REcount: a novel method for highly accurate Illumina sequencing-based quantification
Abstract
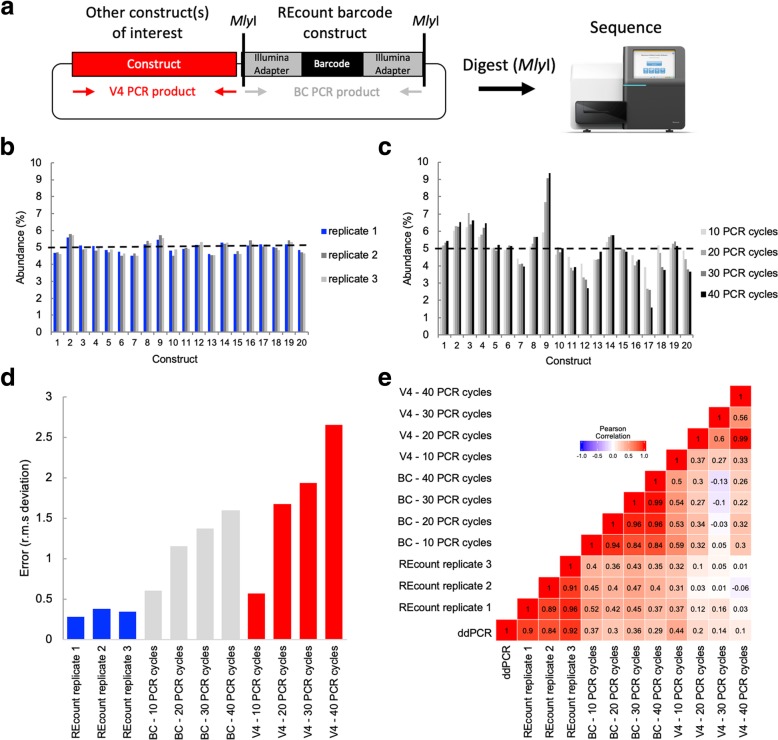
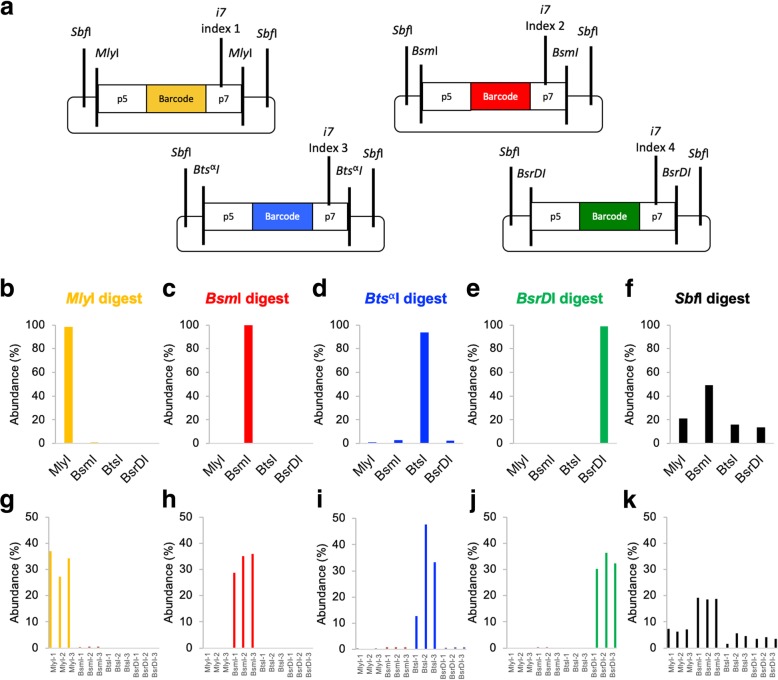
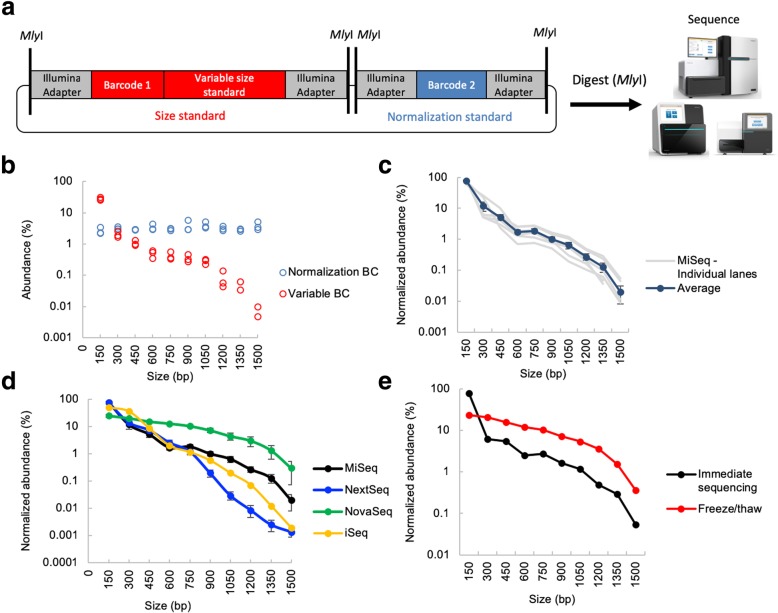
Quantification of DNA sequence tags from engineered constructs such as plasmids, transposons, or other transgenes underlies many functional genomics measurements. Typically, such measurements rely on PCR followed by next-generation sequencing. However, PCR amplification can introduce significant quantitative error. We describe REcount, a novel PCR-free direct counting method. Comparing measurements of defined plasmid pools to droplet digital PCR data demonstrates that REcount is highly accurate and reproducible. We use REcount to provide new insights into clustering biases due to molecule length across different Illumina sequencers and illustrate the impacts on interpretation of next-generation sequencing data and the economics of data generation.
Keywords: ATAC-Seq; DNA library preparation; Genotyping by sequencing; Illumina; Next-generation sequencing; PCR-free; RAD-Seq; RNA-Seq; Size bias.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The REcount PCR-free quantification barcode technology described here is included in US patent application numbers 62/332,879, 62/630,463, and PCT/US17/31271. DMG is the CSO of CoreBiome, Inc. KBB is the COO of CoreBiome, Inc.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
