

STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models
- PMID: 31036082
- PMCID: PMC6489306
- DOI: 10.1186/s40425-019-0573-5
STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models
Abstract
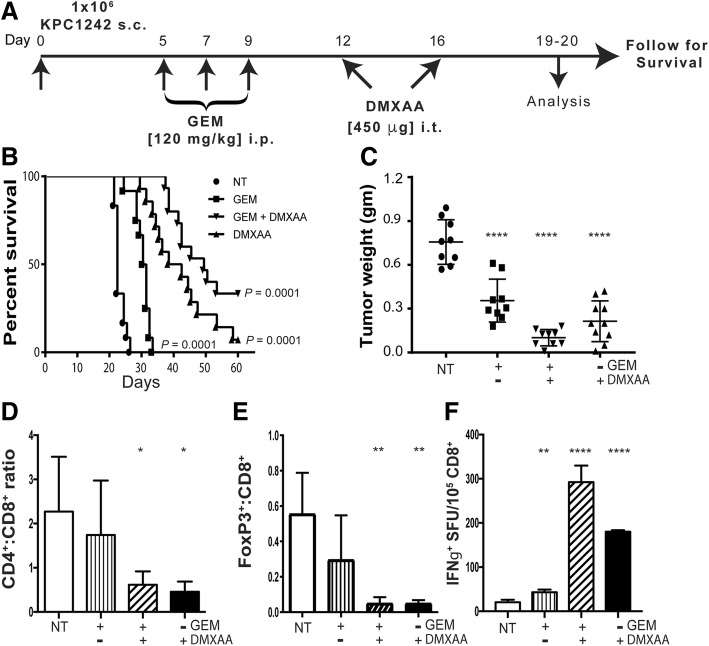
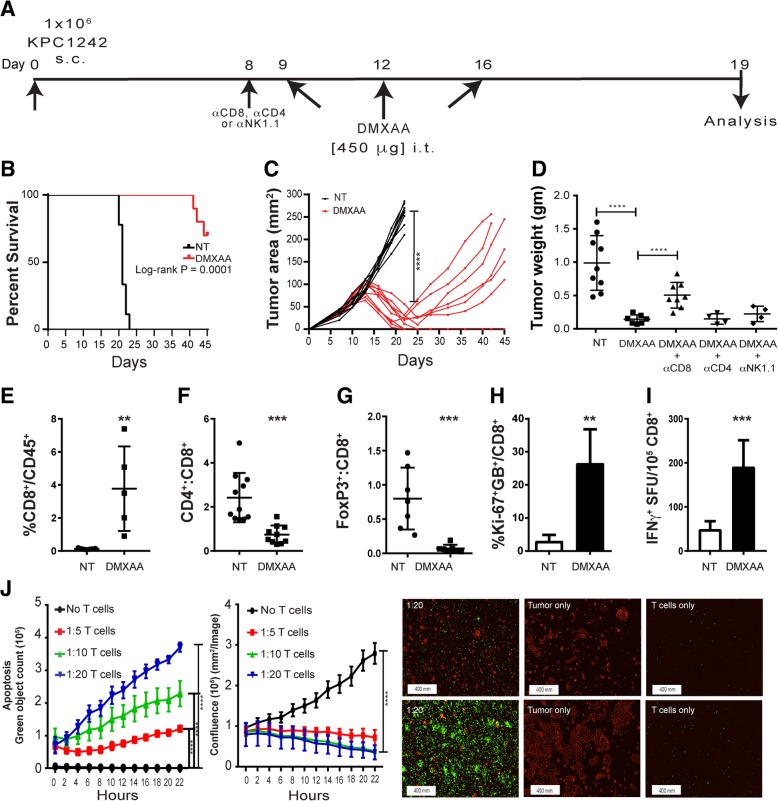
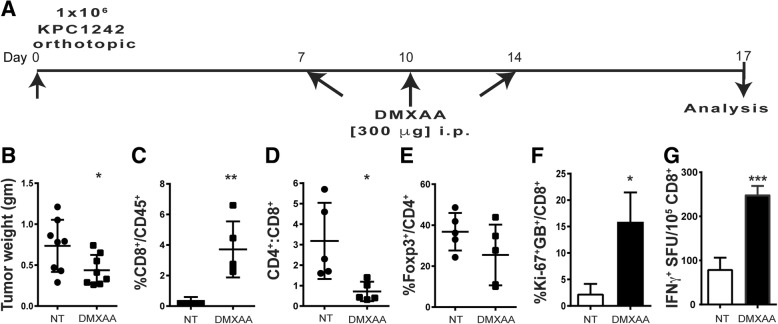
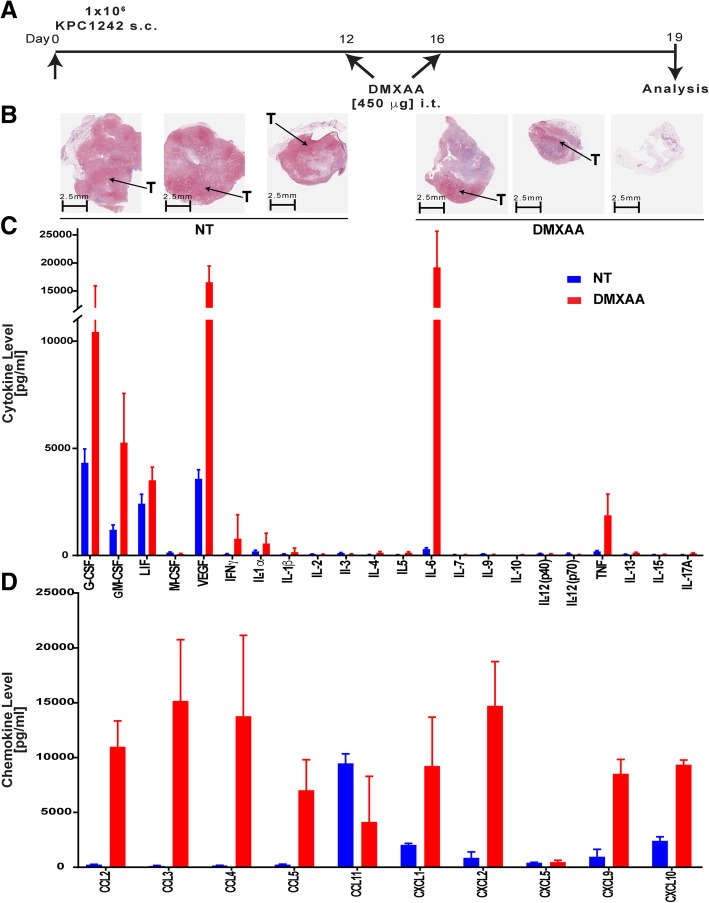
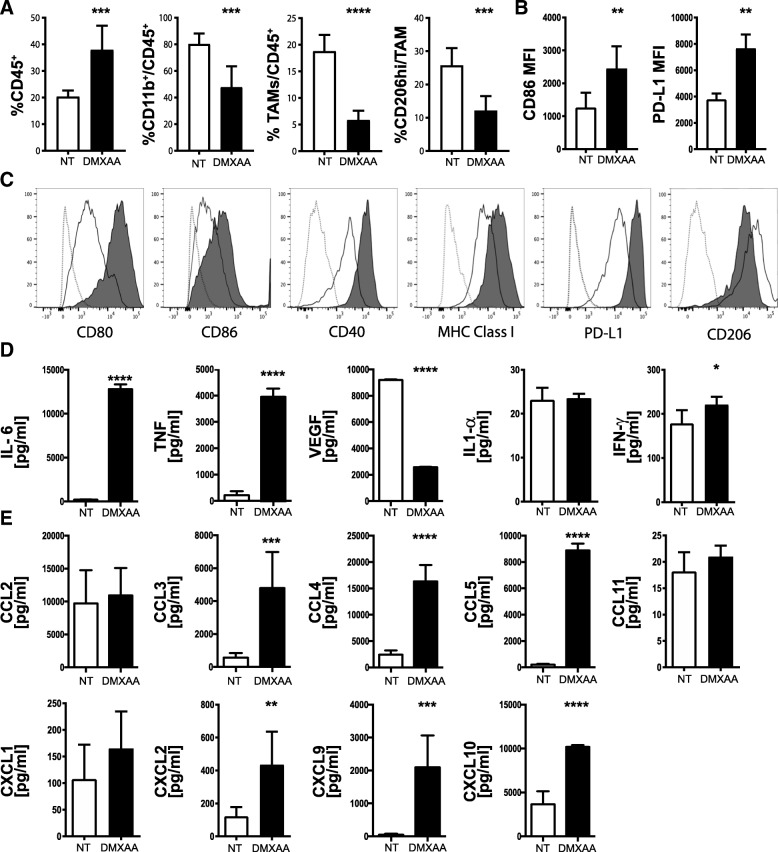
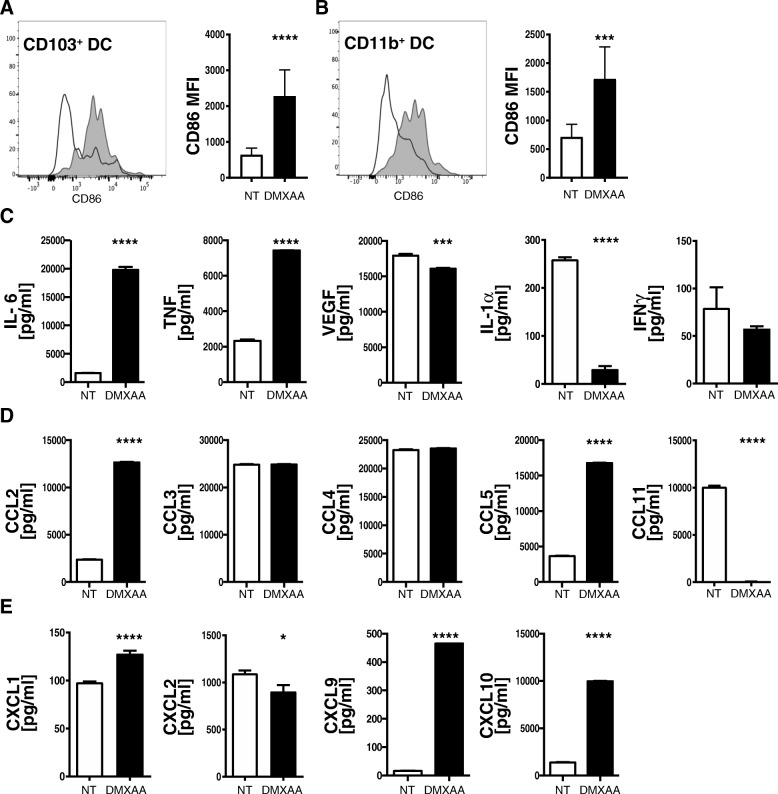
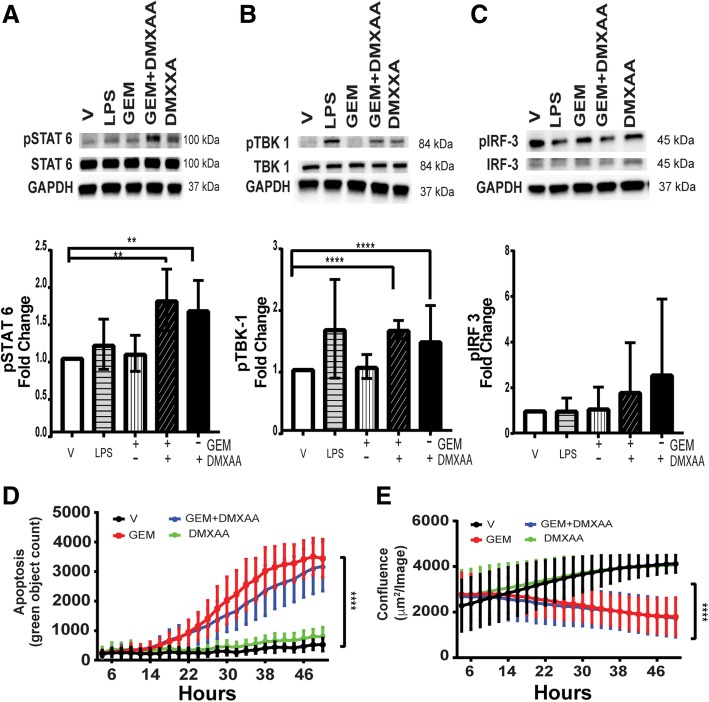
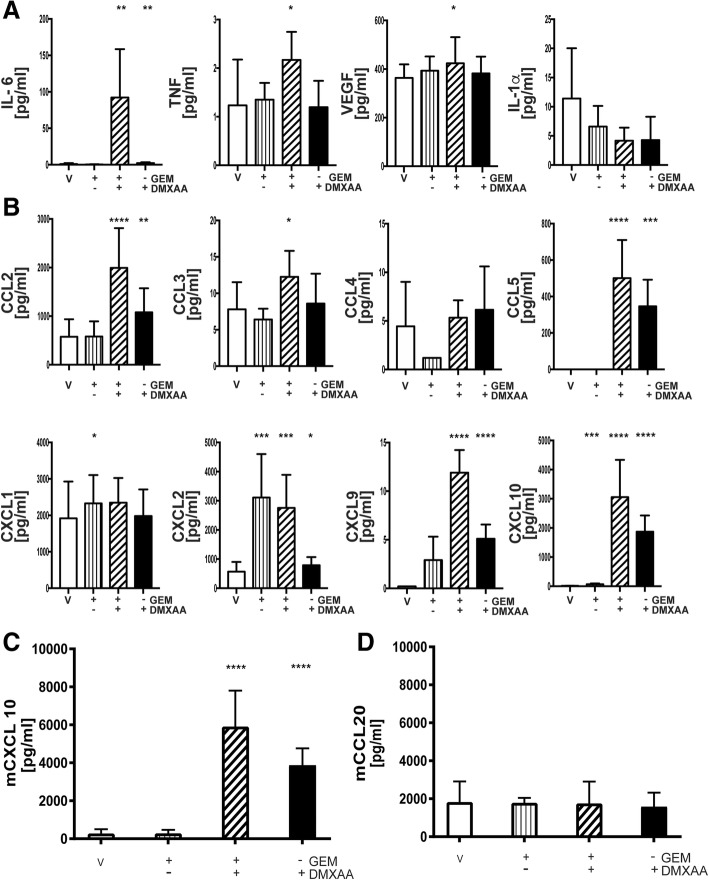
Pancreatic cancer is characterized by an immune suppressive stromal reaction that creates a barrier to therapy. A murine transgenic pancreatic cancer cell line that recapitulates human disease was used to test whether a STimulator of Interferon Genes (STING) agonist could reignite immunologically inert pancreatic tumors. STING agonist treatment potently changed the tumor architecture, altered the immune profile, and increased the survival of tumor-bearing mice. Notably, STING agonist increased numbers and activity of cytotoxic T cells within tumors and decreased levels of suppressive regulatory T cells. Further, STING agonist treatment upregulated costimulatory molecule expression on cross-presenting dendritic cells and reprogrammed immune-suppressive macrophages into immune-activating subtypes. STING agonist promoted the coordinated and differential cytokine production by dendritic cells, macrophages, and pancreatic cancer cells. Cumulatively, these data demonstrate that pancreatic cancer progression is potently inhibited by STING agonist, which reignited immunologically cold pancreatic tumors to promote trafficking and activation of tumor-killing T cells.
Keywords: CXCL10; Cancer proliferation; Cytotoxic T cells; Immune activation; STING agonist; Tumor infiltrating lymphocytes; Tumor selectivity.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
M.B. Dwinell has ownership interests in Protein Foundry, LLC which produces ultra-pure recombinant chemokines for research (
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment.J Immunother Cancer. 2021 Feb;9(2):e001906. doi: 10.1136/jitc-2020-001906. J Immunother Cancer. 2021. PMID: 33526609 Free PMC article.
-
Combined Autophagy Inhibition and Dendritic Cell Recruitment Induces Antitumor Immunity and Enhances Immune Checkpoint Blockade Sensitivity in Pancreatic Cancer.Cancer Res. 2024 Dec 16;84(24):4214-4232. doi: 10.1158/0008-5472.CAN-24-0830. Cancer Res. 2024. PMID: 39288081 Free PMC article.
-
Tumor Cell-Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer.Cancer Res. 2020 Mar 1;80(5):1088-1101. doi: 10.1158/0008-5472.CAN-19-2080. Epub 2020 Jan 8. Cancer Res. 2020. PMID: 31915130 Free PMC article.
-
Killing the "BAD": Challenges for immunotherapy in pancreatic cancer.Biochim Biophys Acta Rev Cancer. 2020 Aug;1874(1):188384. doi: 10.1016/j.bbcan.2020.188384. Epub 2020 Jun 9. Biochim Biophys Acta Rev Cancer. 2020. PMID: 32531324 Review.
-
Prevailing over T cell exhaustion: New developments in the immunotherapy of pancreatic cancer.Cancer Lett. 2016 Oct 10;381(1):259-68. doi: 10.1016/j.canlet.2016.02.057. Epub 2016 Mar 8. Cancer Lett. 2016. PMID: 26968250 Review.
Cited by
-
Trial watch: dendritic cell vaccination for cancer immunotherapy.Oncoimmunology. 2019 Jul 18;8(11):e1638212. doi: 10.1080/2162402X.2019.1638212. eCollection 2019. Oncoimmunology. 2019. PMID: 31646087 Free PMC article. Review.
-
Intratumoral microbiota: roles in cancer initiation, development and therapeutic efficacy.Signal Transduct Target Ther. 2023 Jan 16;8(1):35. doi: 10.1038/s41392-022-01304-4. Signal Transduct Target Ther. 2023. PMID: 36646684 Free PMC article. Review.
-
High glucose-induced STING activation inhibits diabetic wound healing through promoting M1 polarization of macrophages.Cell Death Discov. 2023 Apr 26;9(1):136. doi: 10.1038/s41420-023-01425-x. Cell Death Discov. 2023. PMID: 37100799 Free PMC article.
-
The Role of Chemokines in Orchestrating the Immune Response to Pancreatic Ductal Adenocarcinoma.Cancers (Basel). 2024 Jan 28;16(3):559. doi: 10.3390/cancers16030559. Cancers (Basel). 2024. PMID: 38339310 Free PMC article. Review.
-
Targeting tumour-reprogrammed myeloid cells: the new battleground in cancer immunotherapy.Semin Immunopathol. 2023 Mar;45(2):163-186. doi: 10.1007/s00281-022-00965-1. Epub 2022 Sep 26. Semin Immunopathol. 2023. PMID: 36161514 Free PMC article. Review.
References
-
- Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–289. - PubMed
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. - PubMed
-
- Spanknebel K, Conlon KC. Advances in the surgical management of pancreatic cancer. Cancer J. 2001;7(4):312–323. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials