

Mutations in MAGT1 lead to a glycosylation disorder with a variable phenotype
- PMID: 31036665
- PMCID: PMC6525510
- DOI: 10.1073/pnas.1817815116
Mutations in MAGT1 lead to a glycosylation disorder with a variable phenotype
Abstract
Congenital disorders of glycosylation (CDG) are a group of rare metabolic diseases, due to impaired protein and lipid glycosylation. We identified two patients with defective serum transferrin glycosylation and mutations in the MAGT1 gene. These patients present with a phenotype that is mainly characterized by intellectual and developmental disability. MAGT1 has been described to be a subunit of the oligosaccharyltransferase (OST) complex and more specifically of the STT3B complex. However, it was also claimed that MAGT1 is a magnesium (Mg2+) transporter. So far, patients with mutations in MAGT1 were linked to a primary immunodeficiency, characterized by chronic EBV infections attributed to a Mg2+ homeostasis defect (XMEN). We compared the clinical and cellular phenotype of our two patients to that of an XMEN patient that we recently identified. All three patients have an N-glycosylation defect, as was shown by the study of different substrates, such as GLUT1 and SHBG, demonstrating that the posttranslational glycosylation carried out by the STT3B complex is dysfunctional in all three patients. Moreover, MAGT1 deficiency is associated with an enhanced expression of TUSC3, the homolog protein of MAGT1, pointing toward a compensatory mechanism. Hence, we delineate MAGT1-CDG as a disorder associated with two different clinical phenotypes caused by defects in glycosylation.
Keywords: CDG; XMEN; congenital disorders of glycosylation; oligosaccharyltransferase complex.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
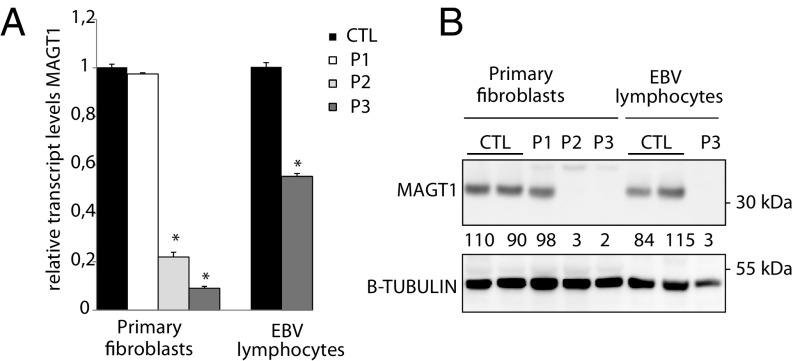
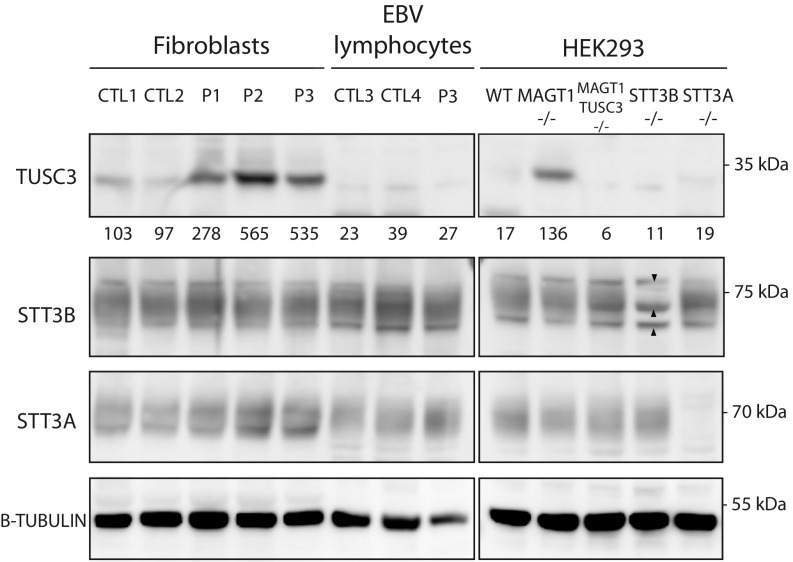
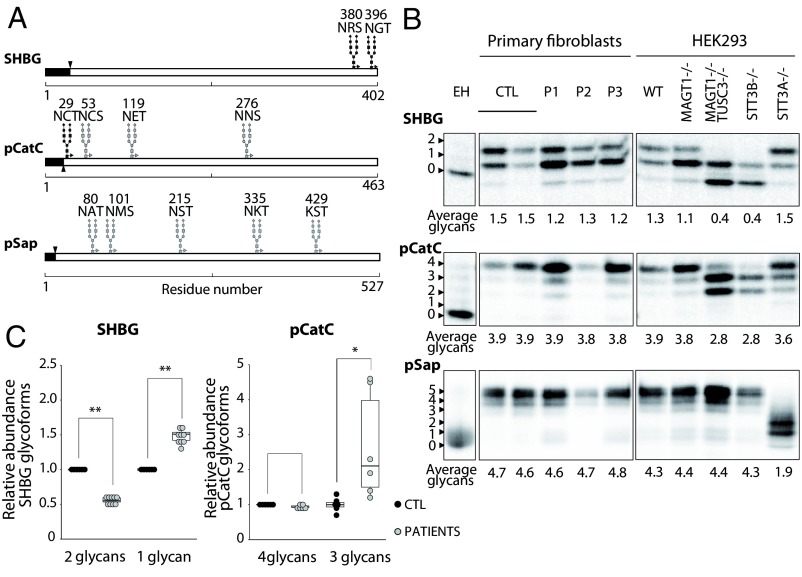

References
-
- Jaeken J, Péanne R. What is new in CDG? J Inherit Metab Dis. 2017;40:569–586. - PubMed
-
- Aebi M. N-linked protein glycosylation in the ER. Biochim Biophys Acta Mol Cell Res. 2013;1833:2430–2437. - PubMed
-
- Kelleher DJ, Karaoglu D, Mandon EC, Gilmore R. Oligosaccharyltransferase isoforms that contain different catalytic STT3 subunits have distinct enzymatic properties. Mol Cell. 2003;12:101–111. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
