

CUT&Tag for efficient epigenomic profiling of small samples and single cells
- PMID: 31036827
- PMCID: PMC6488672
- DOI: 10.1038/s41467-019-09982-5
CUT&Tag for efficient epigenomic profiling of small samples and single cells
Abstract
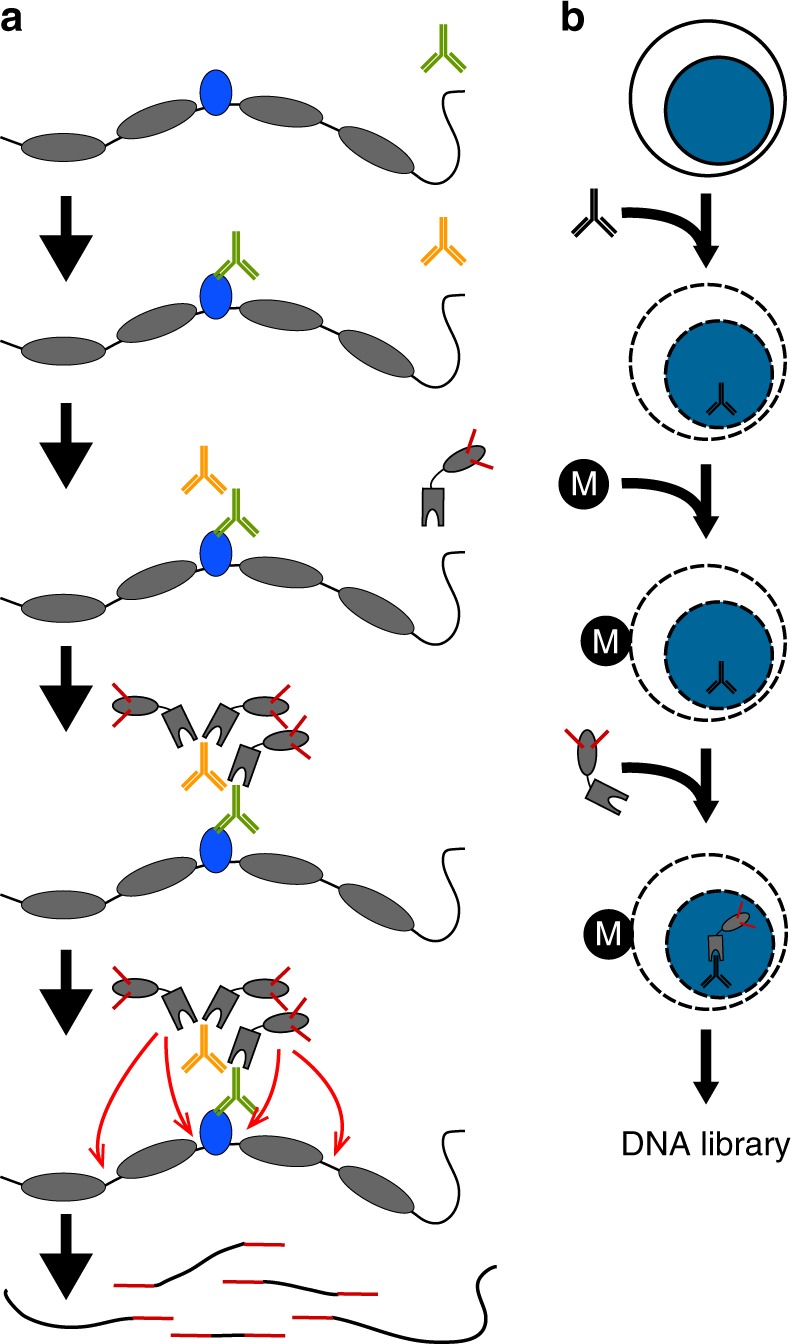
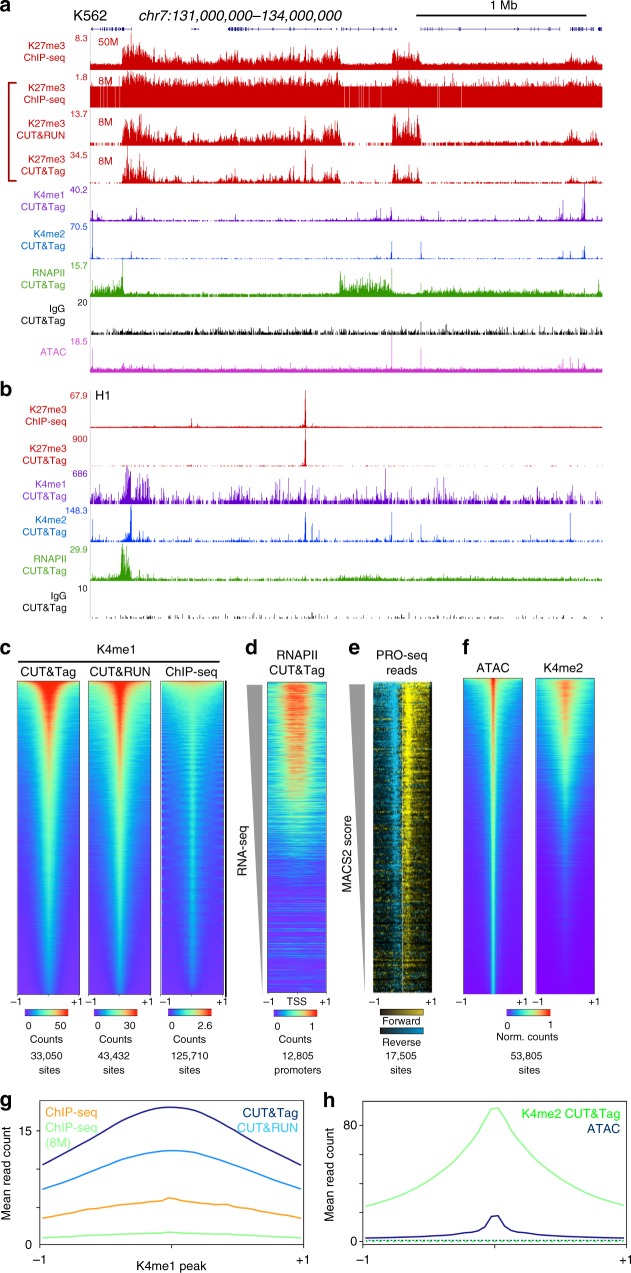
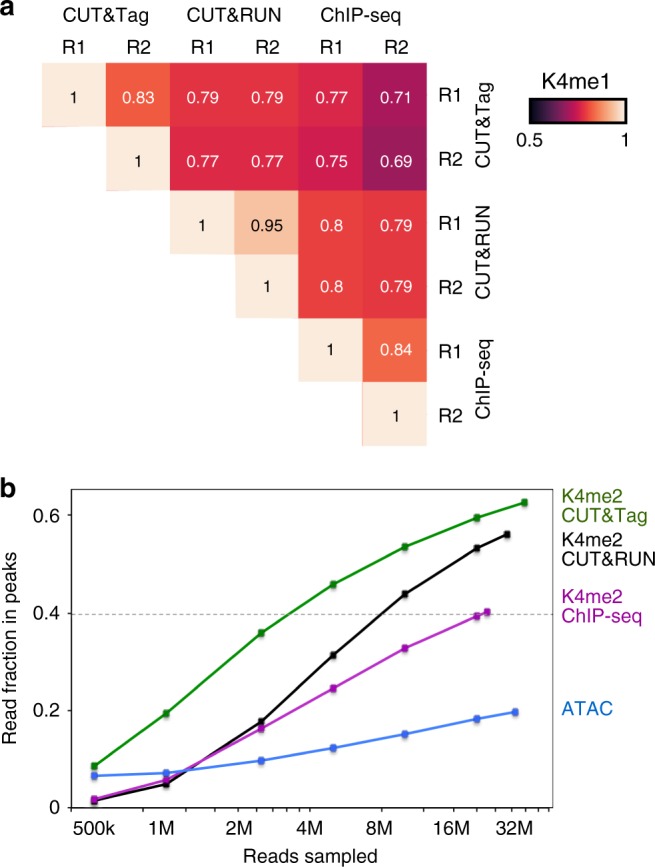
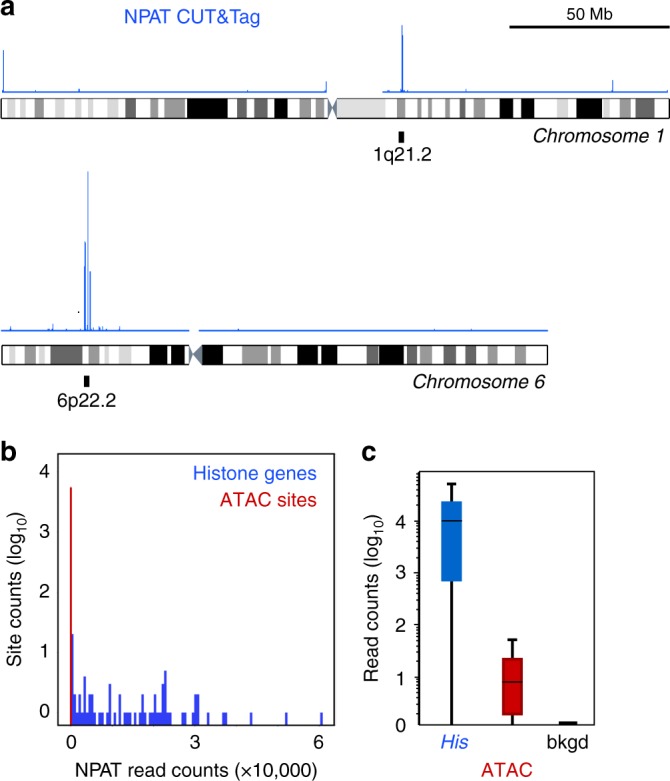
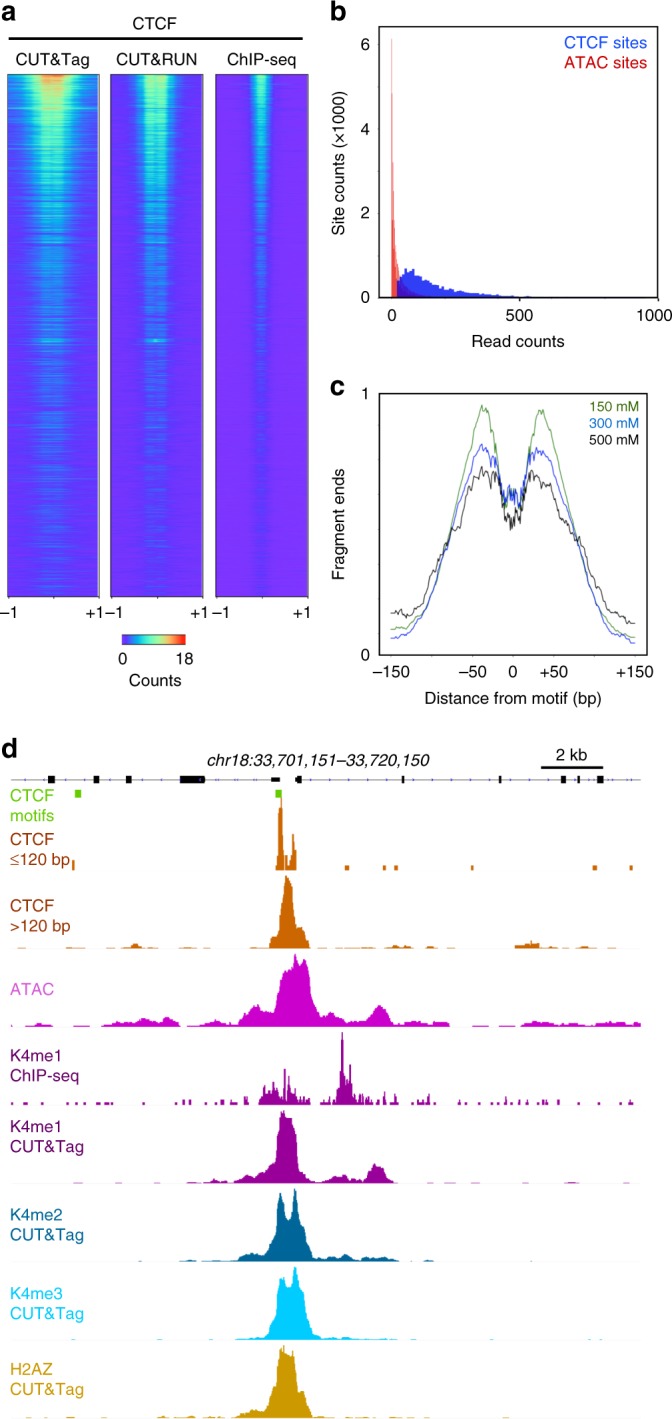
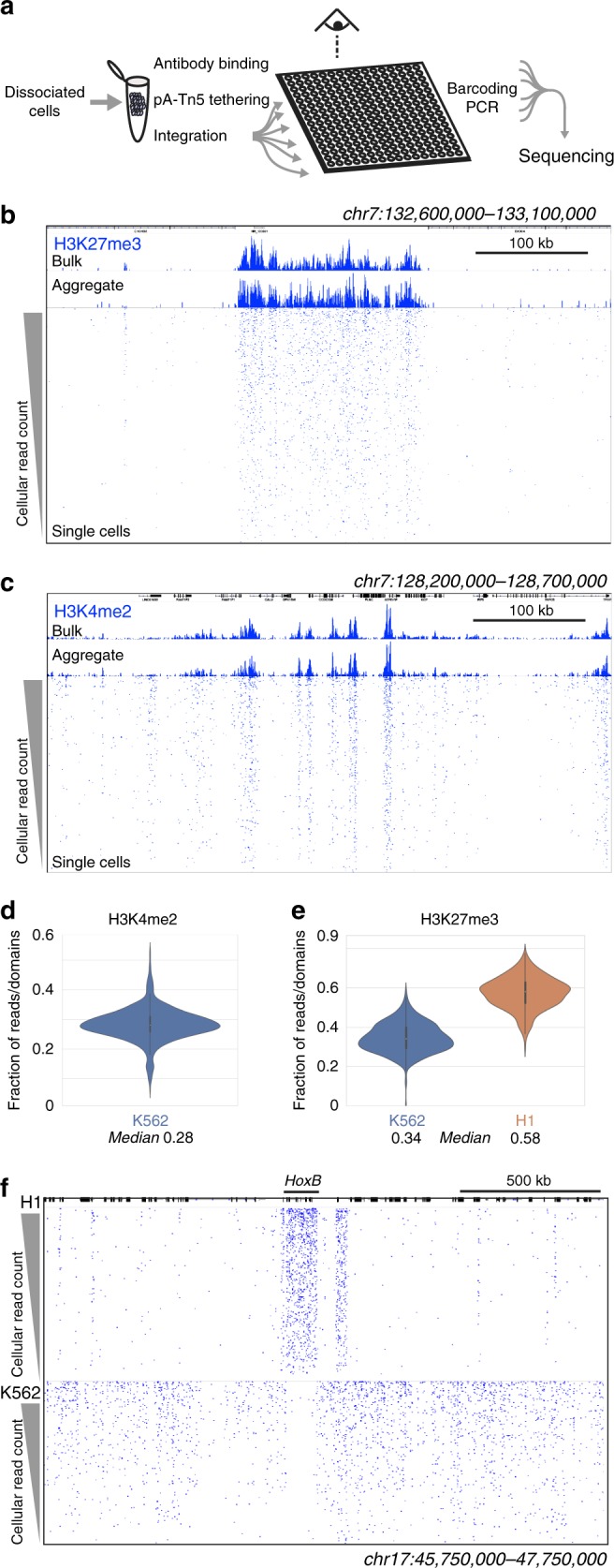
Many chromatin features play critical roles in regulating gene expression. A complete understanding of gene regulation will require the mapping of specific chromatin features in small samples of cells at high resolution. Here we describe Cleavage Under Targets and Tagmentation (CUT&Tag), an enzyme-tethering strategy that provides efficient high-resolution sequencing libraries for profiling diverse chromatin components. In CUT&Tag, a chromatin protein is bound in situ by a specific antibody, which then tethers a protein A-Tn5 transposase fusion protein. Activation of the transposase efficiently generates fragment libraries with high resolution and exceptionally low background. All steps from live cells to sequencing-ready libraries can be performed in a single tube on the benchtop or a microwell in a high-throughput pipeline, and the entire procedure can be performed in one day. We demonstrate the utility of CUT&Tag by profiling histone modifications, RNA Polymerase II and transcription factors on low cell numbers and single cells.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Policastro RA, Zentner GE. Enzymatic methods for genome-wide profiling of protein binding sites. Brief. Funct. Genom. 2018;17:138–145. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
