

Intra- and interpatient evolution of enterovirus D68 analyzed by whole-genome deep sequencing
- PMID: 31037220
- PMCID: PMC6482344
- DOI: 10.1093/ve/vez007
Intra- and interpatient evolution of enterovirus D68 analyzed by whole-genome deep sequencing
Abstract
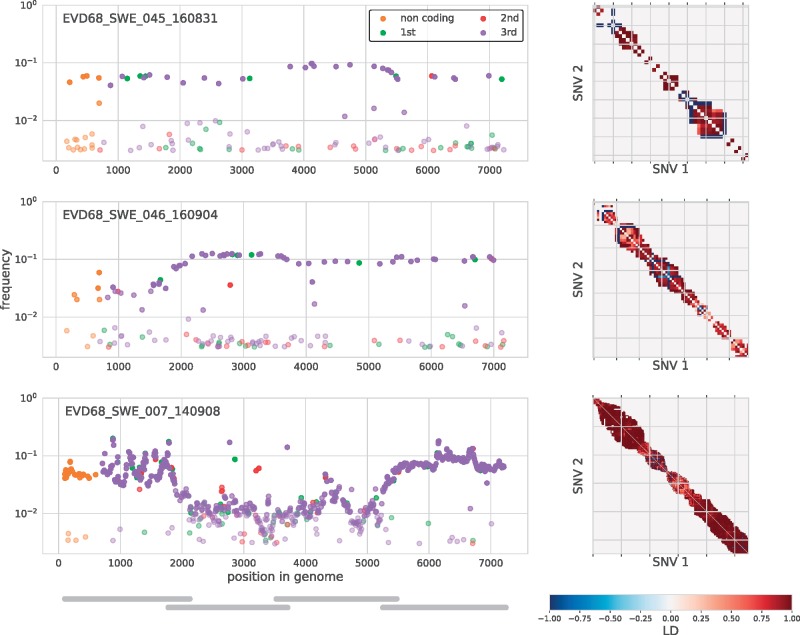
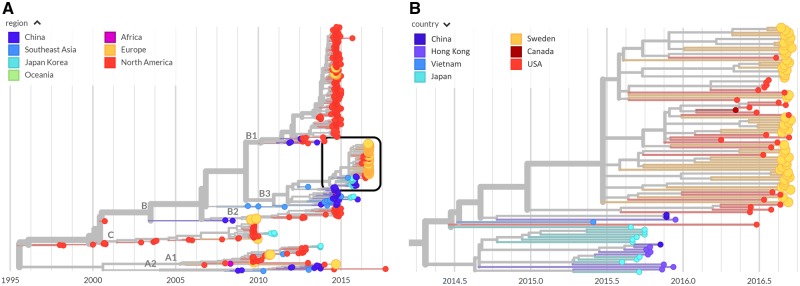
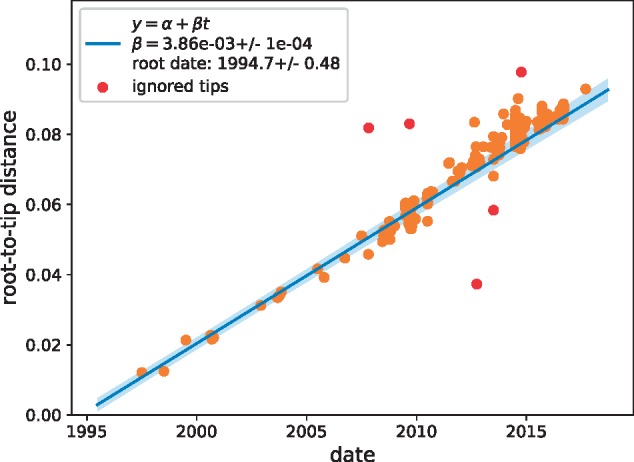
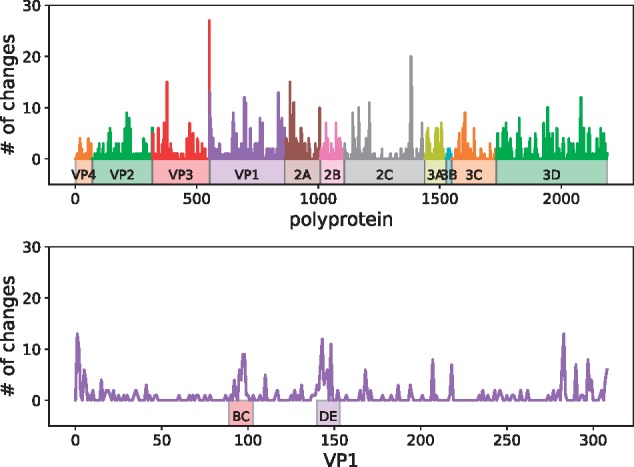
Worldwide outbreaks of enterovirus D68 (EV-D68) in 2014 and 2016 have caused serious respiratory and neurological disease. To investigate diversity, spread, and evolution of EV-D68 we performed near full-length deep sequencing in fifty-four samples obtained in Sweden during the 2014 and 2016 outbreaks. In most samples, intrapatient variability was low and dominated by rare synonymous variants, but three patients showed evidence of dual infections with distinct EV-D68 variants from the same subclade. Interpatient evolution showed a very strong temporal signal, with an evolutionary rate of 0.0039 ± 0.0001 substitutions per site and year. Phylogenetic trees reconstructed from the sequences suggest that EV-D68 was introduced into Stockholm several times during the 2016 outbreak. Putative neutralization targets in the BC and DE loops of the VP1 protein were slightly more diverse within-host and tended to undergo more frequent substitution than other genomic regions. However, evolution in these loops did not appear to have been driven the emergence of the 2016 B3-subclade directly from the 2014 B1-subclade. Instead, the most recent ancestor of both clades was dated to 2009. The study provides a comprehensive description of the intra- and interpatient evolution of EV-D68, including the first report of intrapatient diversity and dual infections. The new data along with publicly available EV-D68 sequences are included in an interactive phylodynamic analysis on nextstrain.org/enterovirus/d68 to facilitate timely EV-D68 tracking in the future.
Keywords: enterovirus; interpatient evolution; intrahost evolution; whole-genome deep sequencing.
Figures
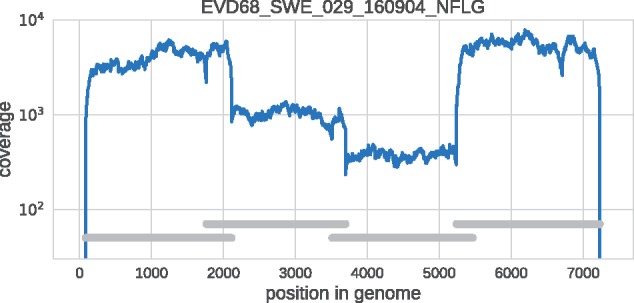
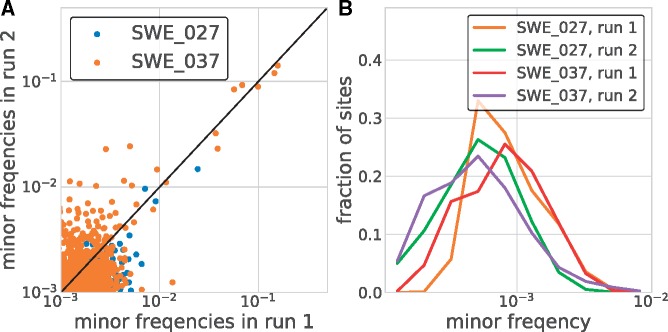
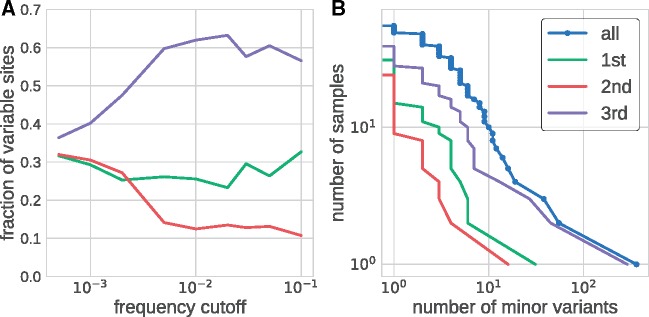
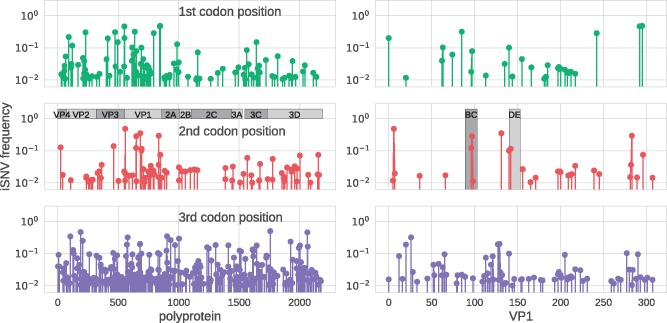
References
-
- Barnadas C. et al. (2017) ‘An Enhanced Enterovirus Surveillance System Allows Identification and Characterization of Rare and Emerging Respiratory Enteroviruses in Denmark, 2015-16’, Journal of Clinical Virology, 93: 40–4. - PubMed
-
- Dai W. et al. (2018) ‘A Virus-like Particle Vaccine Confers Protection against Enterovirus D68 Lethal Challenge in Mice’, Vaccine, 36: 653–9. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
