

High-Fidelity Nanopore Sequencing of Ultra-Short DNA Targets
- PMID: 31038923
- PMCID: PMC6533607
- DOI: 10.1021/acs.analchem.9b00856
High-Fidelity Nanopore Sequencing of Ultra-Short DNA Targets
Abstract
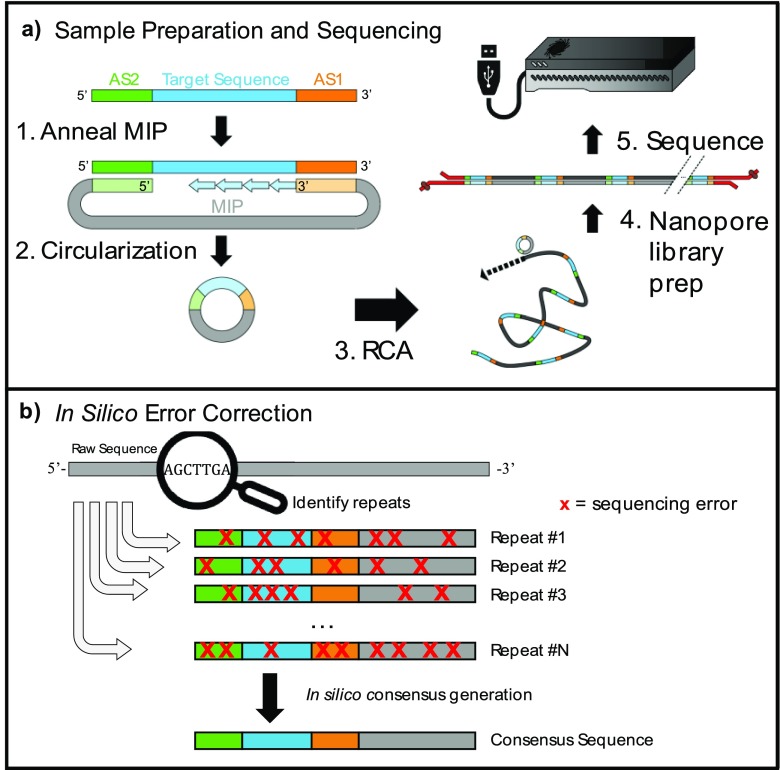
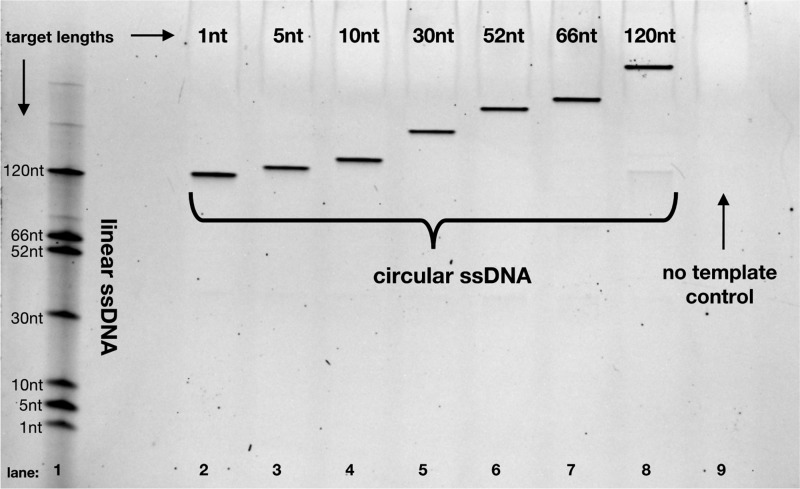
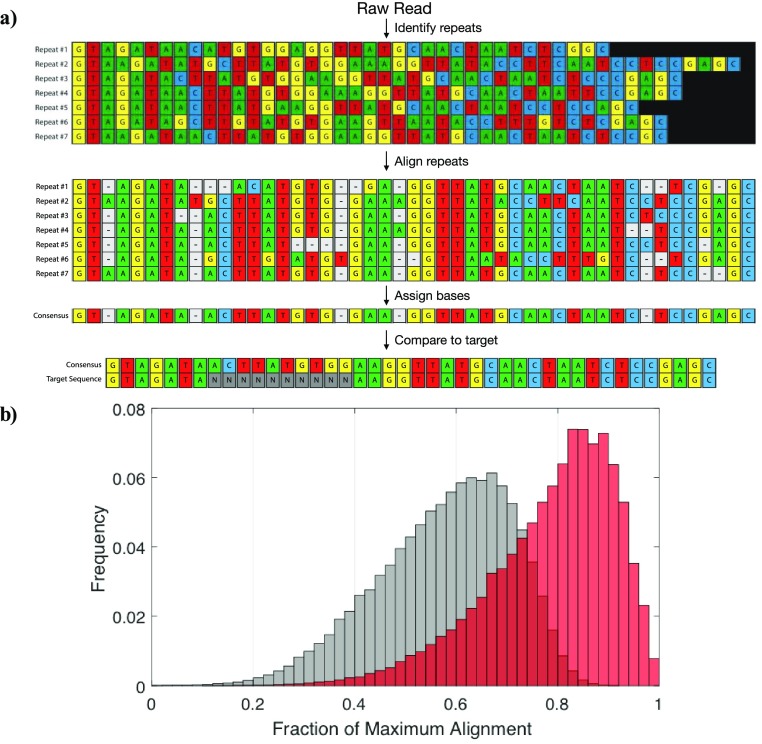
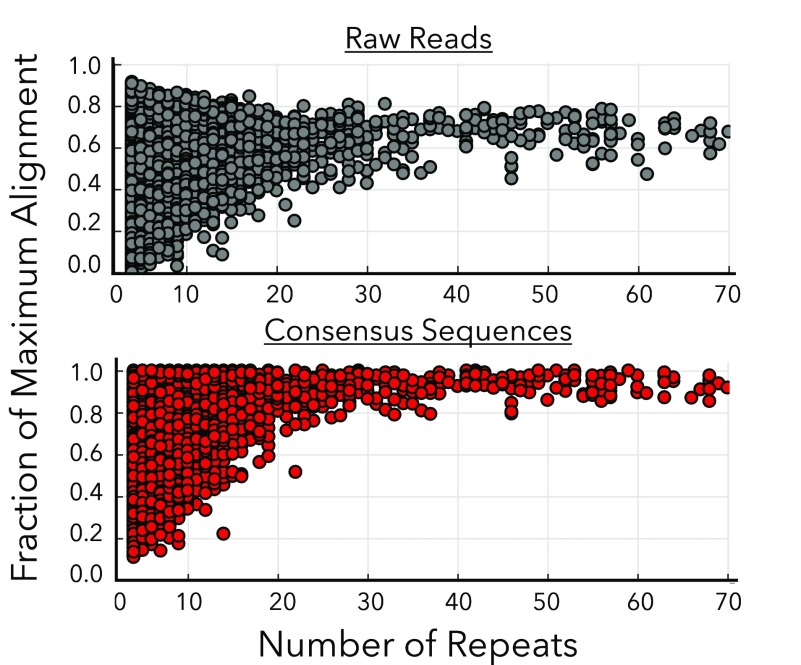
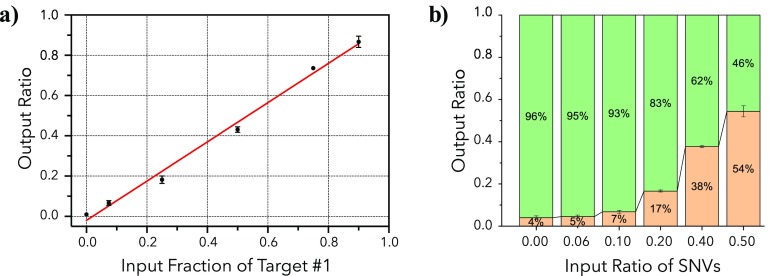
Nanopore sequencing offers a portable and affordable alternative to sequencing-by-synthesis methods but suffers from lower accuracy and cannot sequence ultrashort DNA. This puts applications such as molecular diagnostics based on the analysis of cell-free DNA or single-nucleotide variants (SNVs) out of reach. To overcome these limitations, we report a nanopore-based sequencing strategy in which short target sequences are first circularized and then amplified via rolling-circle amplification to produce long stretches of concatemeric repeats. After sequencing on the Oxford Nanopore Technologies MinION platform, the resulting repeat sequences can be aligned to produce a highly accurate consensus that reduces the high error-rate present in the individual repeats. Using this approach, we demonstrate for the first time the ability to obtain unbiased and accurate nanopore data for target DNA sequences <100 bp. Critically, this approach is sensitive enough to achieve SNV discrimination in mixtures of sequences and even enables quantitative detection of specific variants present at ratios of <10%. Our method is simple, cost-effective, and only requires well-established processes. It therefore expands the utility of nanopore sequencing for molecular diagnostics and other applications, especially in resource-limited settings.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
Rolling circle reverse transcription enables high fidelity nanopore sequencing of small RNA.PLoS One. 2022 Oct 10;17(10):e0275471. doi: 10.1371/journal.pone.0275471. eCollection 2022. PLoS One. 2022. PMID: 36215256 Free PMC article.
-
Custom-Primed Rolling Circle Amplicons for Highly Accurate Nanopore Sequencing.Small Methods. 2025 Jun;9(6):e2401416. doi: 10.1002/smtd.202401416. Epub 2025 Mar 3. Small Methods. 2025. PMID: 40025906
-
Genome assembly using Nanopore-guided long and error-free DNA reads.BMC Genomics. 2015 Apr 20;16(1):327. doi: 10.1186/s12864-015-1519-z. BMC Genomics. 2015. PMID: 25927464 Free PMC article.
-
Potential applications of nanopore sequencing for forensic analysis.Forensic Sci Rev. 2020 Jan;32(1):23-54. Forensic Sci Rev. 2020. PMID: 32007927 Review.
-
Nanopore sequencing: Review of potential applications in functional genomics.Dev Growth Differ. 2019 Jun;61(5):316-326. doi: 10.1111/dgd.12608. Epub 2019 Apr 29. Dev Growth Differ. 2019. PMID: 31037722 Review.
Cited by
-
Rapid in situ identification of biological specimens via DNA amplicon sequencing using miniaturized laboratory equipment.Nat Protoc. 2022 Jun;17(6):1415-1443. doi: 10.1038/s41596-022-00682-x. Epub 2022 Apr 11. Nat Protoc. 2022. PMID: 35411044 Review.
-
Opportunities and challenges in long-read sequencing data analysis.Genome Biol. 2020 Feb 7;21(1):30. doi: 10.1186/s13059-020-1935-5. Genome Biol. 2020. PMID: 32033565 Free PMC article. Review.
-
In vivo hypermutation and continuous evolution.Nat Rev Methods Primers. 2022;2:37. doi: 10.1038/s43586-022-00130-w. Epub 2022 May 19. Nat Rev Methods Primers. 2022. PMID: 37073402 Free PMC article. No abstract available.
-
Long-read human genome sequencing and its applications.Nat Rev Genet. 2020 Oct;21(10):597-614. doi: 10.1038/s41576-020-0236-x. Epub 2020 Jun 5. Nat Rev Genet. 2020. PMID: 32504078 Free PMC article. Review.
-
Genetic Biomonitoring and Biodiversity Assessment Using Portable Sequencing Technologies: Current Uses and Future Directions.Genes (Basel). 2019 Oct 29;10(11):858. doi: 10.3390/genes10110858. Genes (Basel). 2019. PMID: 31671909 Free PMC article. Review.
References
-
- Mitchell P. S.; Parkin R. K.; Kroh E. M.; Fritz B. R.; Wyman S. K.; Pogosova-Agadjanyan E. L.; Peterson A.; Noteboom J.; Briant K. C. O.; Allen A.; et al. Circulating MicroRNAs as Stable Blood-Based Markers for Cancer Detection. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (30), 10513–10518. 10.1073/pnas.0804549105. - DOI - PMC - PubMed
-
- Krishnakumar R.; Sinha A.; Bird S. W.; Jayamohan H.; Edwards H. S.; Schoeniger J. S.; Patel K. D.; Branda S. S.; Bartsch M. S. Systematic and Stochastic Influences on the Performance of the MinION Nanopore Sequencer across a Range of Nucleotide Bias. Sci. Rep. 2018, 8 (1), 1–13. 10.1038/s41598-018-21484-w. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
