

Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report
- PMID: 31039256
- PMCID: PMC6536849
- DOI: 10.1093/brain/awz099
Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report
Erratum in
-
Erratum.Brain. 2019 Jul 1;142(7):e37. doi: 10.1093/brain/awz163. Brain. 2019. PMID: 31147685 Free PMC article. No abstract available.
Abstract
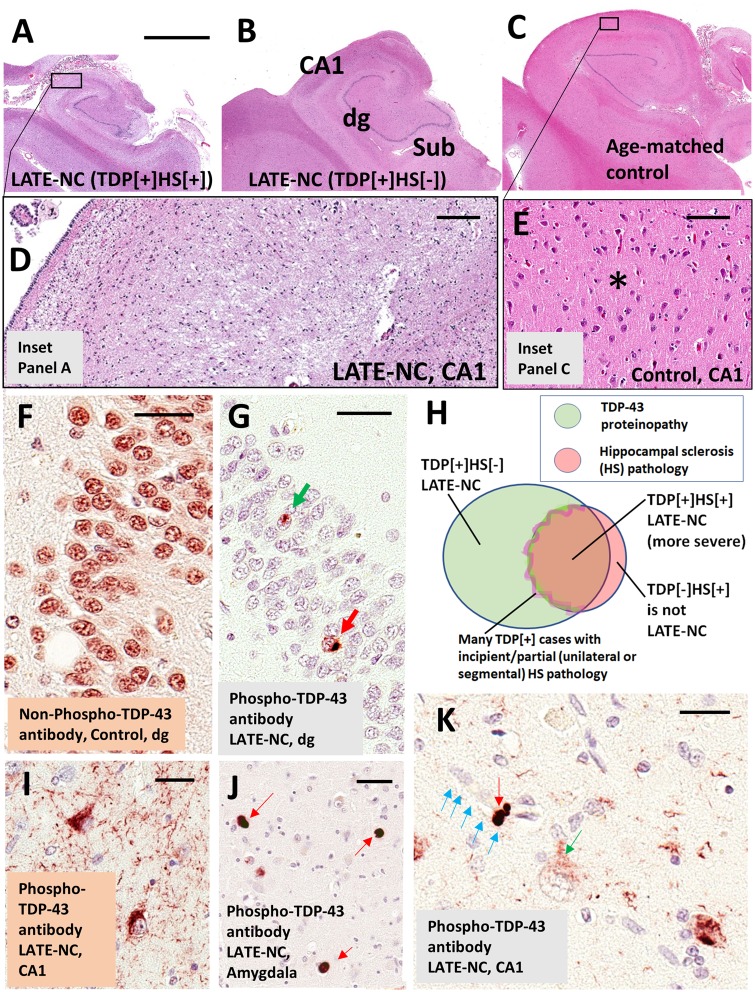
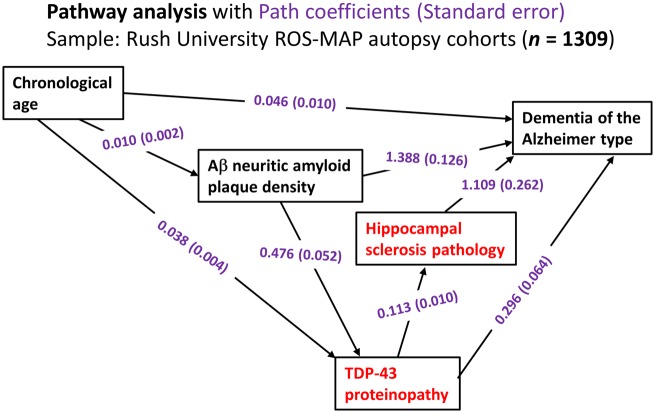
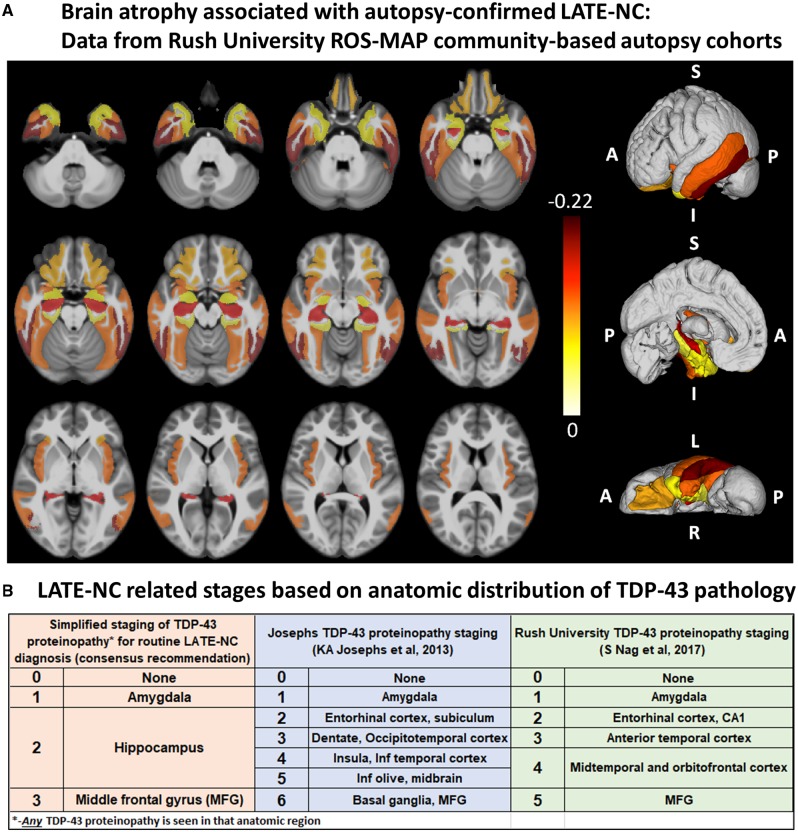
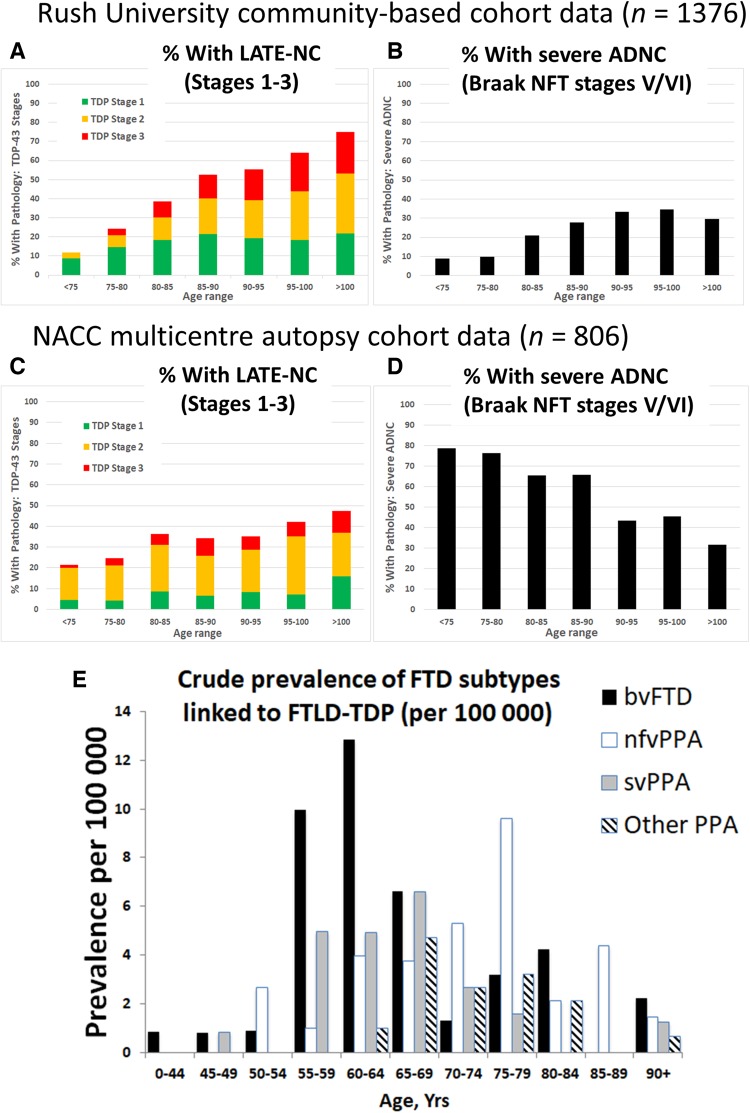
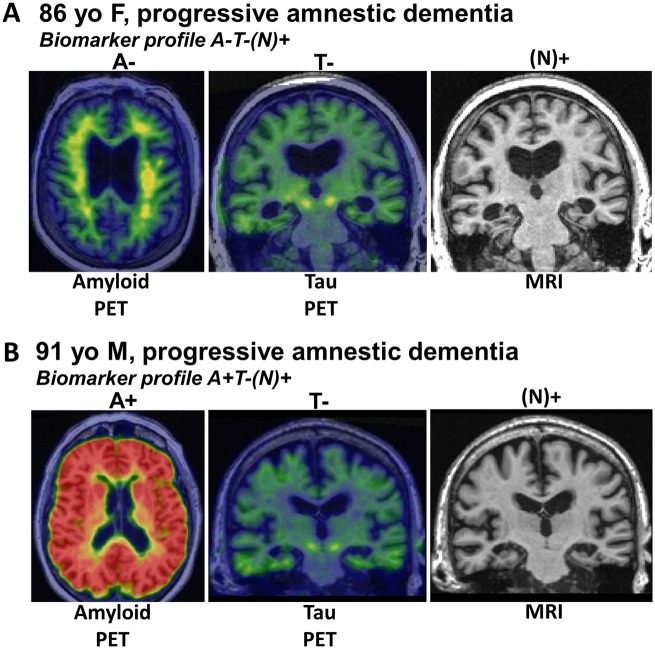
We describe a recently recognized disease entity, limbic-predominant age-related TDP-43 encephalopathy (LATE). LATE neuropathological change (LATE-NC) is defined by a stereotypical TDP-43 proteinopathy in older adults, with or without coexisting hippocampal sclerosis pathology. LATE-NC is a common TDP-43 proteinopathy, associated with an amnestic dementia syndrome that mimicked Alzheimer's-type dementia in retrospective autopsy studies. LATE is distinguished from frontotemporal lobar degeneration with TDP-43 pathology based on its epidemiology (LATE generally affects older subjects), and relatively restricted neuroanatomical distribution of TDP-43 proteinopathy. In community-based autopsy cohorts, ∼25% of brains had sufficient burden of LATE-NC to be associated with discernible cognitive impairment. Many subjects with LATE-NC have comorbid brain pathologies, often including amyloid-β plaques and tauopathy. Given that the 'oldest-old' are at greatest risk for LATE-NC, and subjects of advanced age constitute a rapidly growing demographic group in many countries, LATE has an expanding but under-recognized impact on public health. For these reasons, a working group was convened to develop diagnostic criteria for LATE, aiming both to stimulate research and to promote awareness of this pathway to dementia. We report consensus-based recommendations including guidelines for diagnosis and staging of LATE-NC. For routine autopsy workup of LATE-NC, an anatomically-based preliminary staging scheme is proposed with TDP-43 immunohistochemistry on tissue from three brain areas, reflecting a hierarchical pattern of brain involvement: amygdala, hippocampus, and middle frontal gyrus. LATE-NC appears to affect the medial temporal lobe structures preferentially, but other areas also are impacted. Neuroimaging studies demonstrated that subjects with LATE-NC also had atrophy in the medial temporal lobes, frontal cortex, and other brain regions. Genetic studies have thus far indicated five genes with risk alleles for LATE-NC: GRN, TMEM106B, ABCC9, KCNMB2, and APOE. The discovery of these genetic risk variants indicate that LATE shares pathogenetic mechanisms with both frontotemporal lobar degeneration and Alzheimer's disease, but also suggests disease-specific underlying mechanisms. Large gaps remain in our understanding of LATE. For advances in prevention, diagnosis, and treatment, there is an urgent need for research focused on LATE, including in vitro and animal models. An obstacle to clinical progress is lack of diagnostic tools, such as biofluid or neuroimaging biomarkers, for ante-mortem detection of LATE. Development of a disease biomarker would augment observational studies seeking to further define the risk factors, natural history, and clinical features of LATE, as well as eventual subject recruitment for targeted therapies in clinical trials.
Keywords: FTLD; MRI; PET; SNAP; epidemiology.
© The Author(s) (2019). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Comment in
-
Piecing together a consensus on a TDP43-related dementia syndrome.Nat Rev Neurol. 2019 Jul;15(7):367. doi: 10.1038/s41582-019-0207-z. Nat Rev Neurol. 2019. PMID: 31110344 No abstract available.
-
Limbic-predominant age-related TDP-43 encephalopathy (LATE).Brain. 2019 Aug 1;142(8):e42. doi: 10.1093/brain/awz184. Brain. 2019. PMID: 31243434 No abstract available.
-
LATE to the PART-y.Brain. 2019 Sep 1;142(9):e47. doi: 10.1093/brain/awz224. Brain. 2019. PMID: 31359030 Free PMC article. No abstract available.
-
Selected cryptic exons accumulate in hippocampal cell nuclei in Alzheimer's disease with and without associated TDP-43 proteinopathy.Brain. 2020 Mar 1;143(3):e20. doi: 10.1093/brain/awaa013. Brain. 2020. PMID: 32016361 No abstract available.
-
Reply: Selected cryptic exons accumulate in hippocampal cell nuclei in Alzheimer's disease with and without associated TDP-43 proteinopathy.Brain. 2020 Mar 1;143(3):e21. doi: 10.1093/brain/awaa014. Brain. 2020. PMID: 32016396 Free PMC article. No abstract available.
References
-
- Ala TA, Beh GO, Frey WH 2nd. Pure hippocampal sclerosis: a rare cause of dementia mimicking Alzheimer’s disease. Neurology 2000; 54: 843–8. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 AG028303/AG/NIA NIH HHS/United States
- R01 AG042210/AG/NIA NIH HHS/United States
- P50 AG005131/AG/NIA NIH HHS/United States
- U01 AG016976/AG/NIA NIH HHS/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- UF1 AG057707/AG/NIA NIH HHS/United States
- P30 AG010161/AG/NIA NIH HHS/United States
- P50 AG025688/AG/NIA NIH HHS/United States
- P50 AG047366/AG/NIA NIH HHS/United States
- P30 AG028383/AG/NIA NIH HHS/United States
- R01 AG057187/AG/NIA NIH HHS/United States
- R56 AG057191/AG/NIA NIH HHS/United States
- R01 AG041851/AG/NIA NIH HHS/United States
- P30 AG010124/AG/NIA NIH HHS/United States
- G1100540/MRC_/Medical Research Council/United Kingdom
- P50 AG005142/AG/NIA NIH HHS/United States
- R01 AG061111/AG/NIA NIH HHS/United States
- P50 AG016574/AG/NIA NIH HHS/United States
- U24 AG072122/AG/NIA NIH HHS/United States
- P01 AG017586/AG/NIA NIH HHS/United States
- UF1 AG053983/AG/NIA NIH HHS/United States
- R01 AG054449/AG/NIA NIH HHS/United States
- R35 NS097261/NS/NINDS NIH HHS/United States
- R01 NS094003/NS/NINDS NIH HHS/United States
- P30 AG049638/AG/NIA NIH HHS/United States
- P30 AG012300/AG/NIA NIH HHS/United States
- R01 AG034374/AG/NIA NIH HHS/United States
- R01 AG037491/AG/NIA NIH HHS/United States
- P01 AG003949/AG/NIA NIH HHS/United States
- R37 AG011378/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
