

Homozygous TRPV4 mutation causes congenital distal spinal muscular atrophy and arthrogryposis
- PMID: 31041394
- PMCID: PMC6454305
- DOI: 10.1212/NXG.0000000000000312
Homozygous TRPV4 mutation causes congenital distal spinal muscular atrophy and arthrogryposis
Abstract
Objective: To identify the genetic cause of disease in a form of congenital spinal muscular atrophy and arthrogryposis (CSMAA).
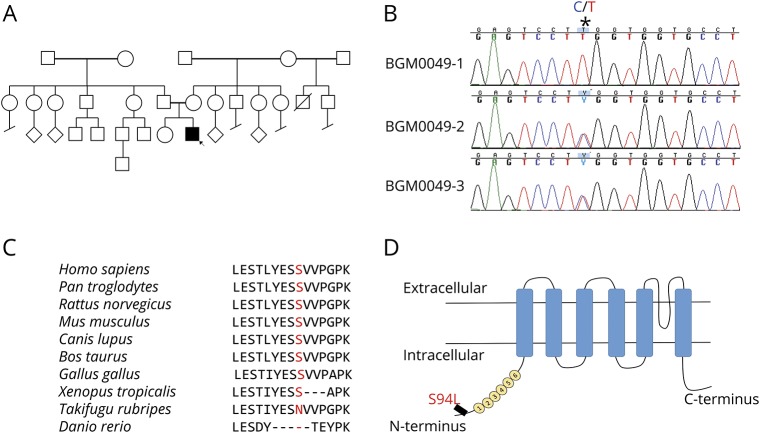
Methods: A 2-year-old boy was diagnosed with arthrogryposis multiplex congenita, severe skeletal abnormalities, torticollis, vocal cord paralysis, and diminished lower limb movement. Whole-exome sequencing (WES) was performed on the proband and family members. In silico modeling of protein structure and heterologous protein expression and cytotoxicity assays were performed to validate pathogenicity of the identified variant.
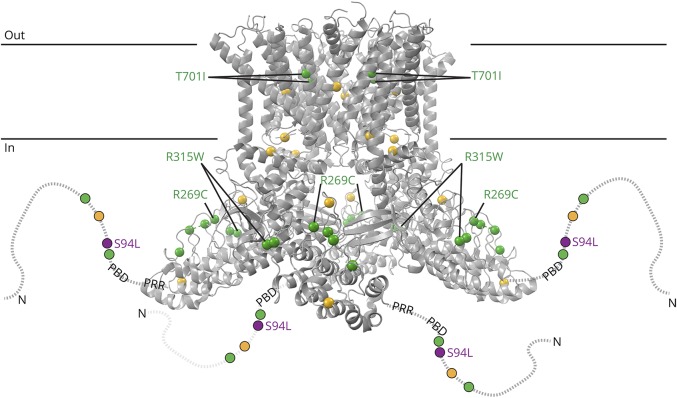
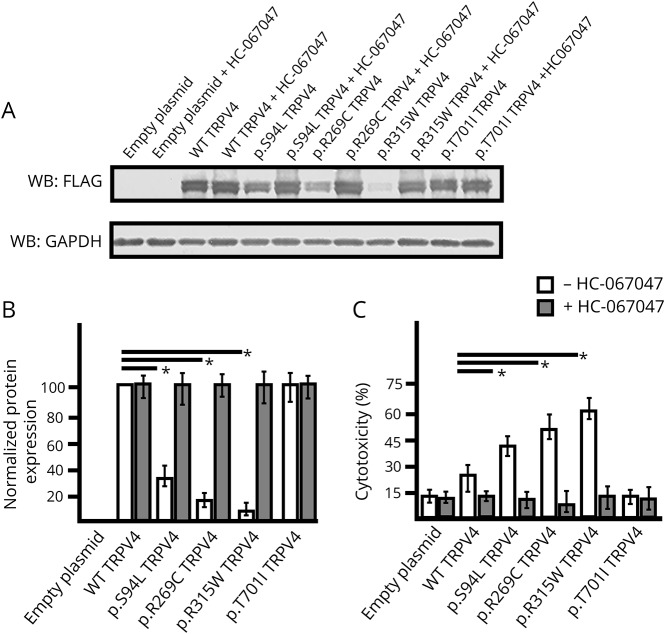
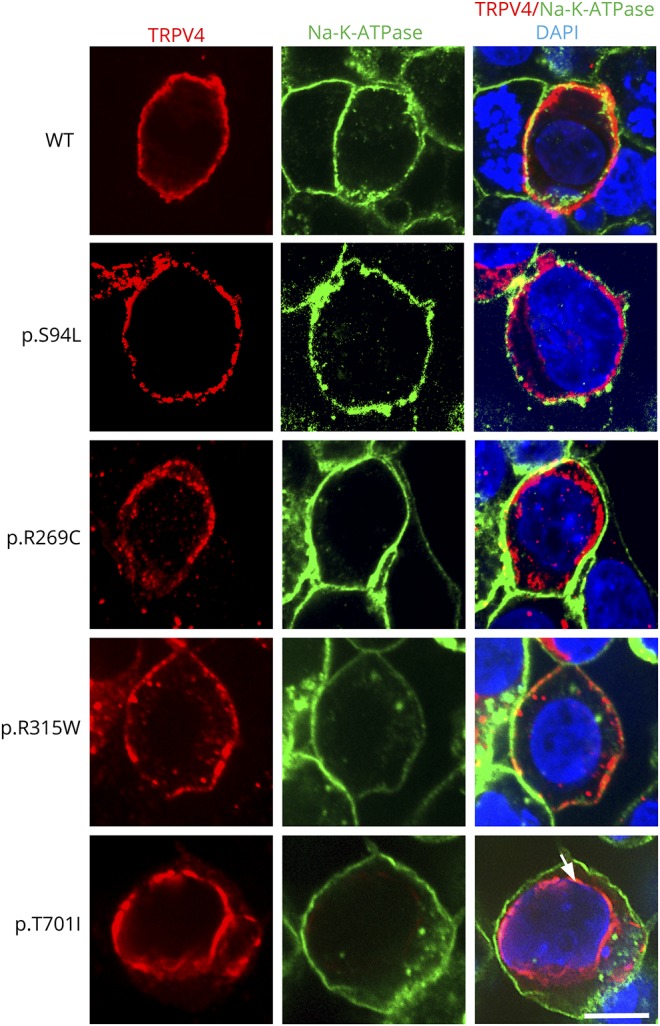
Results: WES revealed a homozygous mutation in the TRPV4 gene (c.281C>T; p.S94L). The identification of a recessive mutation in TRPV4 extends the spectrum of mutations in recessive forms of the TRPV4-associated disease. p.S94L and other previously identified TRPV4 variants in different protein domains were compared in structural modeling and functional studies. In silico structural modeling suggests that the p.S94L mutation is in the disordered N-terminal region proximal to important regulatory binding sites for phosphoinositides and for PACSIN3, which could lead to alterations in trafficking and/or channel sensitivity. Functional studies by Western blot and immunohistochemical analysis show that p.S94L increased TRPV4 activity-based cytotoxicity and resultant decreased TRPV4 expression levels, therefore involves a gain-of-function mechanism.
Conclusions: This study identifies a novel homozygous mutation in TRPV4 as a cause of the recessive form of CSMAA.
Figures
Comment in
-
Long-term benefits of nusinersen in later-onset spinal muscular atrophy.Nat Rev Neurol. 2019 Jul;15(7):368-369. doi: 10.1038/s41582-019-0202-4. Nat Rev Neurol. 2019. PMID: 31065075 No abstract available.
References
-
- Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol 2013;9:562–571. - PubMed
-
- Pareyson D, Saveri P, Pisciotta C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr Opin Neurol 2017;30:471–480. - PubMed
-
- Fawcett KA, Murphy SM, Polke JM, et al. . Comprehensive analysis of the TRPV4 gene in a large series of inherited neuropathies and controls. J Neurol Neurosurg Psychiatry 2012;83:1204–1209. - PubMed
LinkOut - more resources
Full Text Sources