

Childhood cerebellar tumours mirror conserved fetal transcriptional programs
- PMID: 31043743
- PMCID: PMC6675628
- DOI: 10.1038/s41586-019-1158-7
Childhood cerebellar tumours mirror conserved fetal transcriptional programs
Abstract
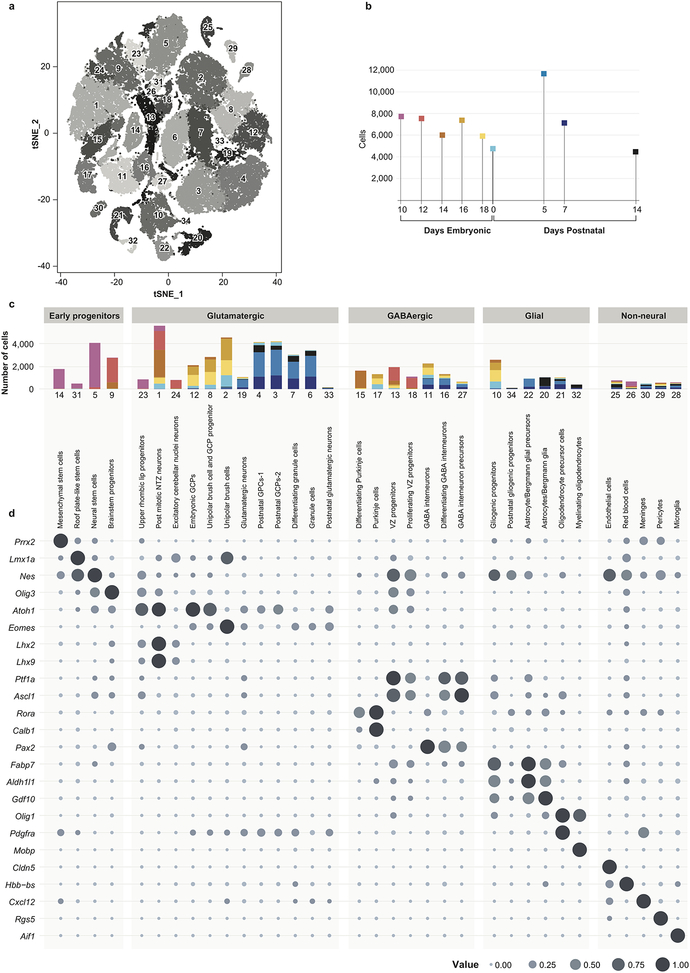
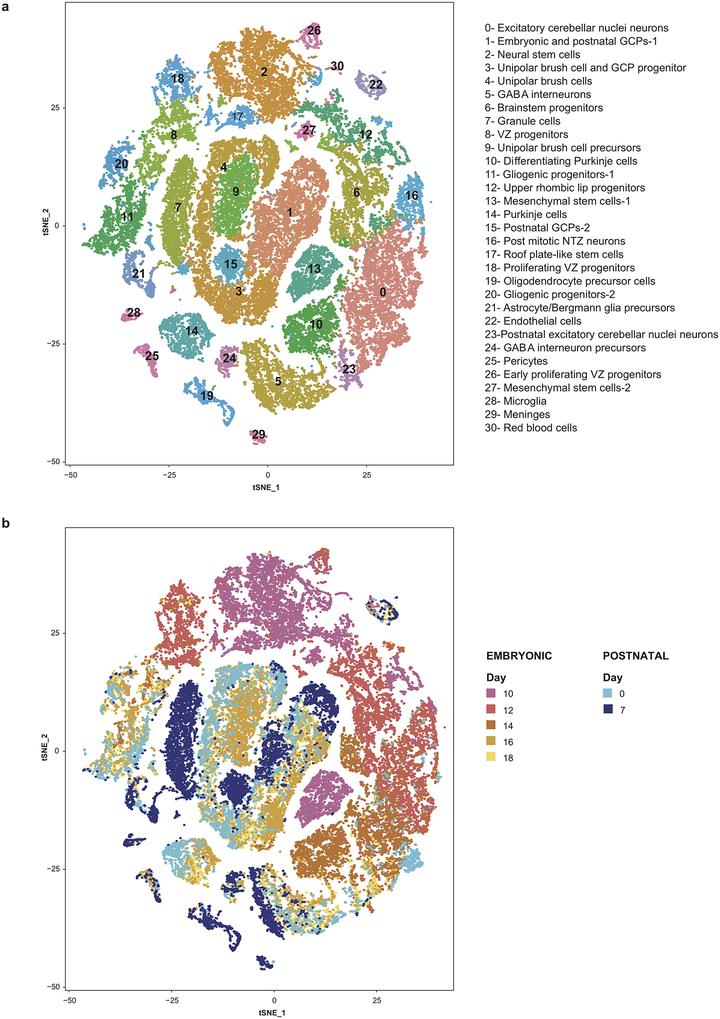
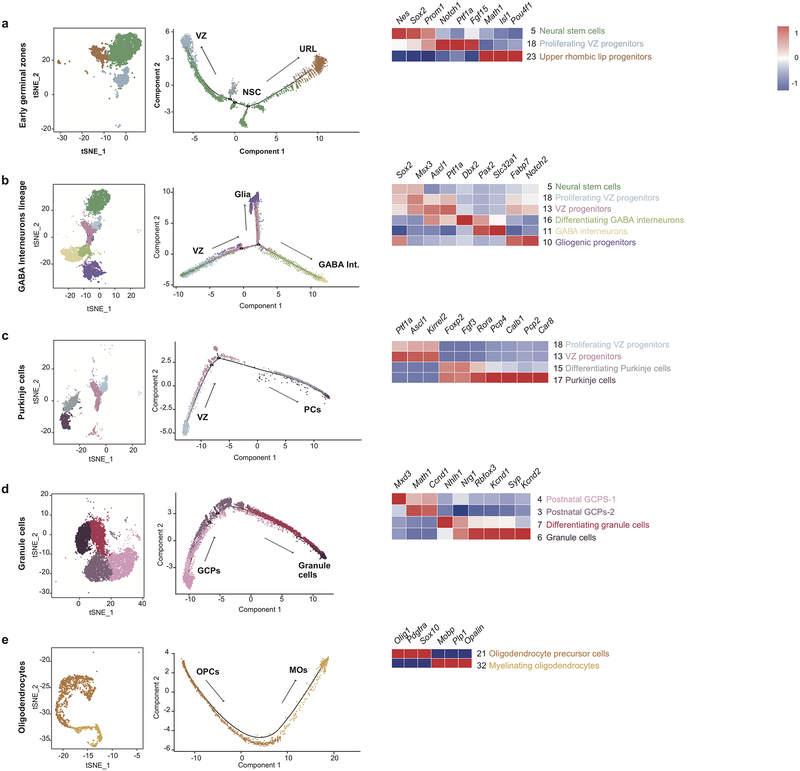
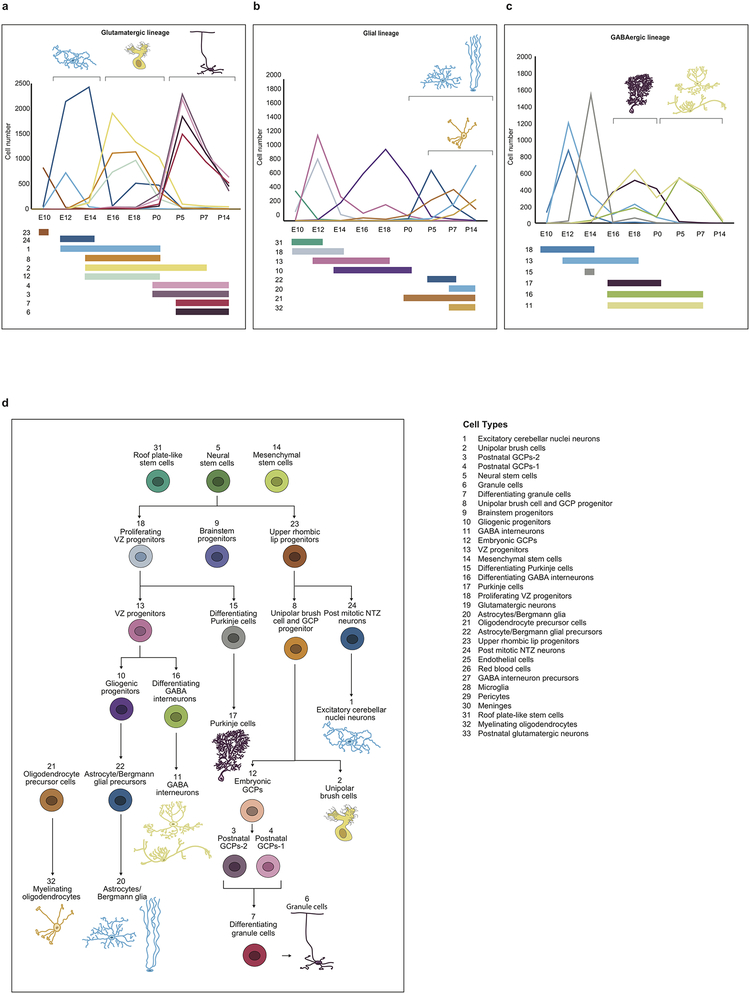
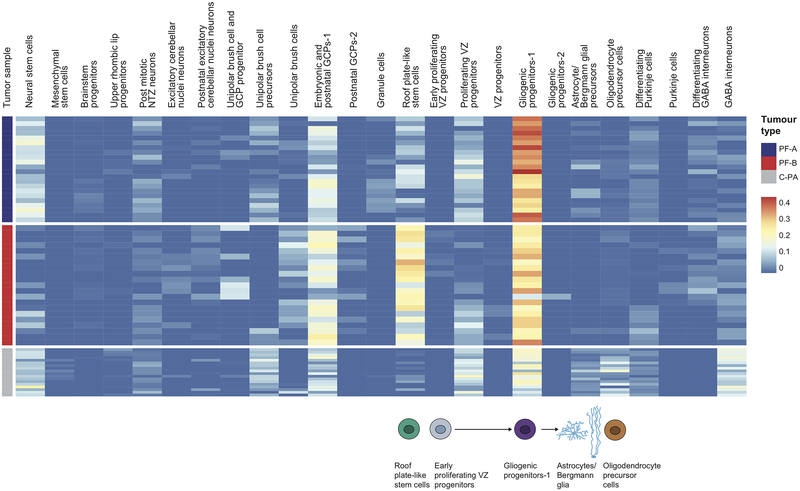
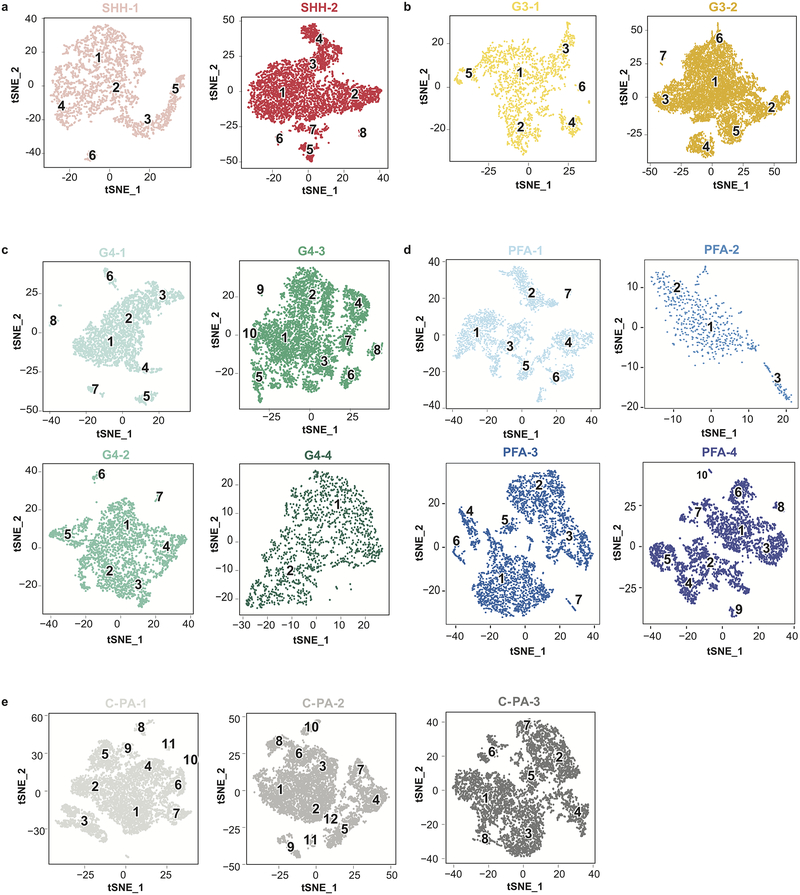
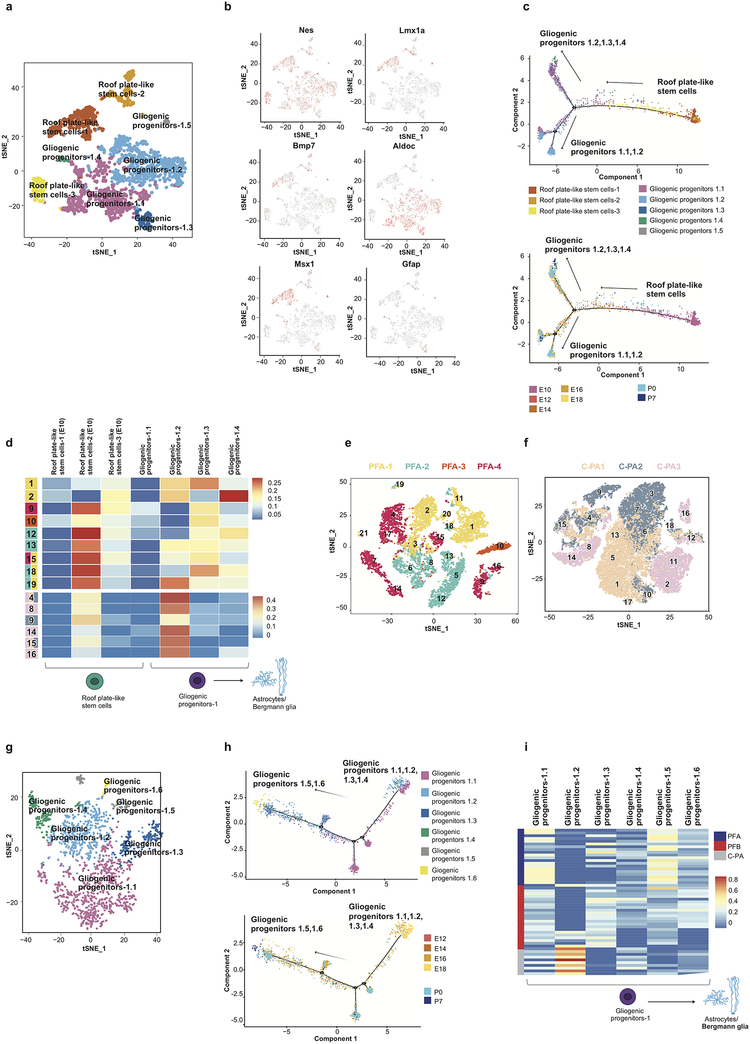
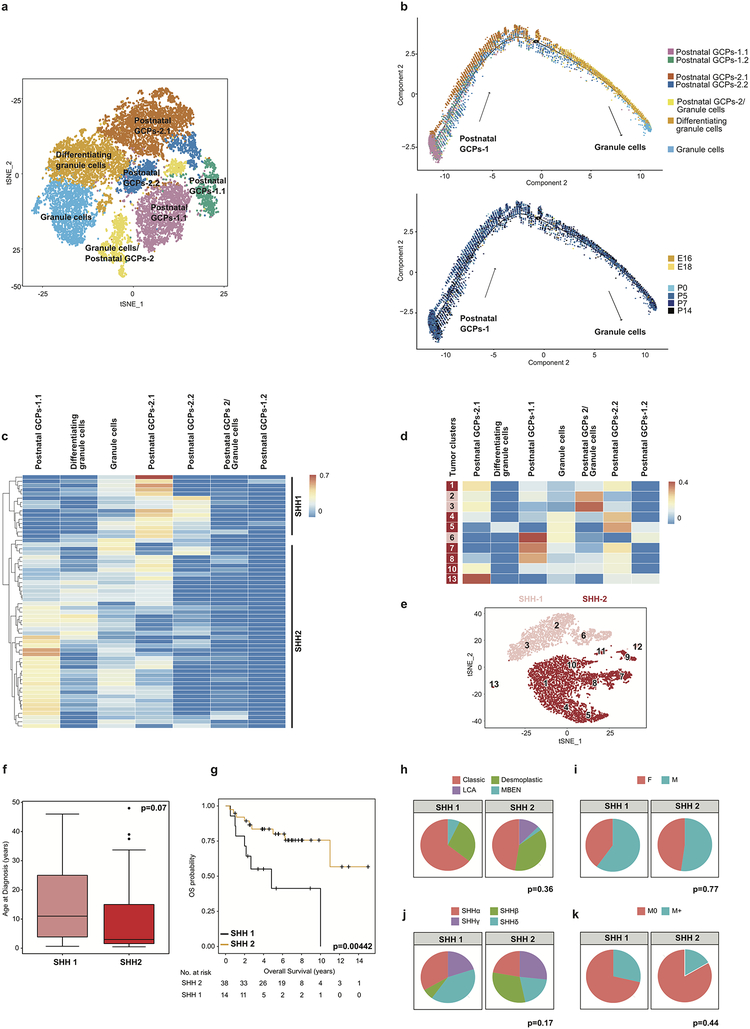
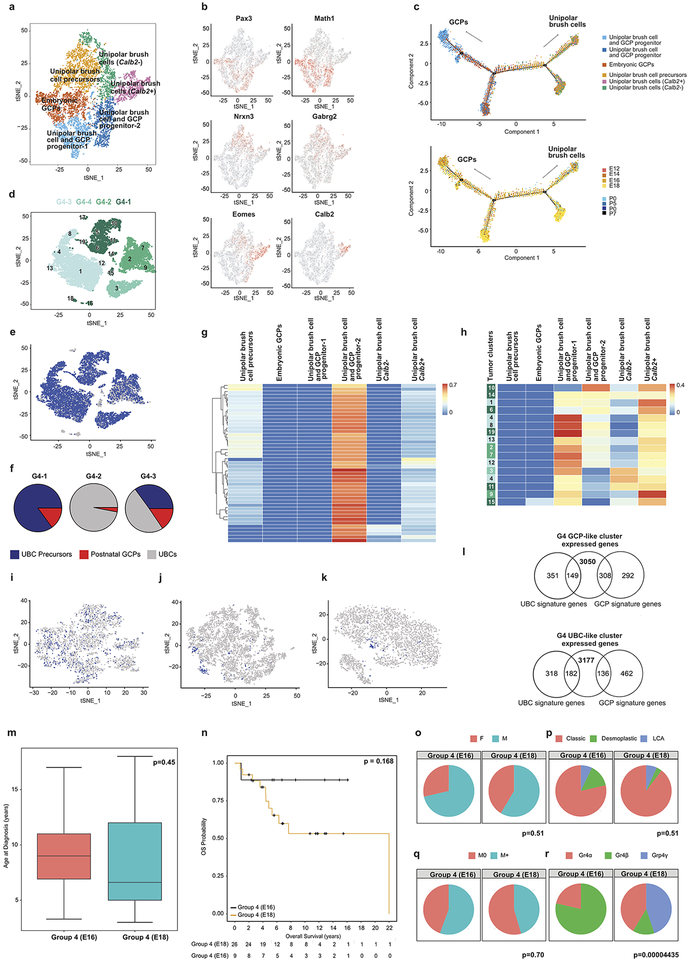
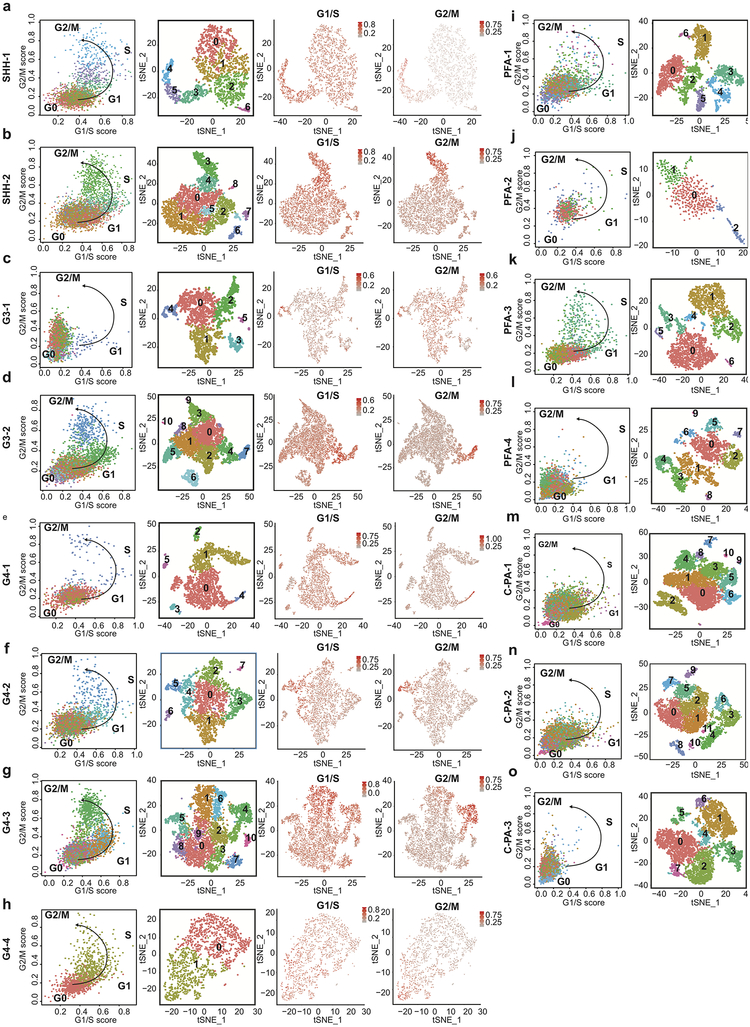
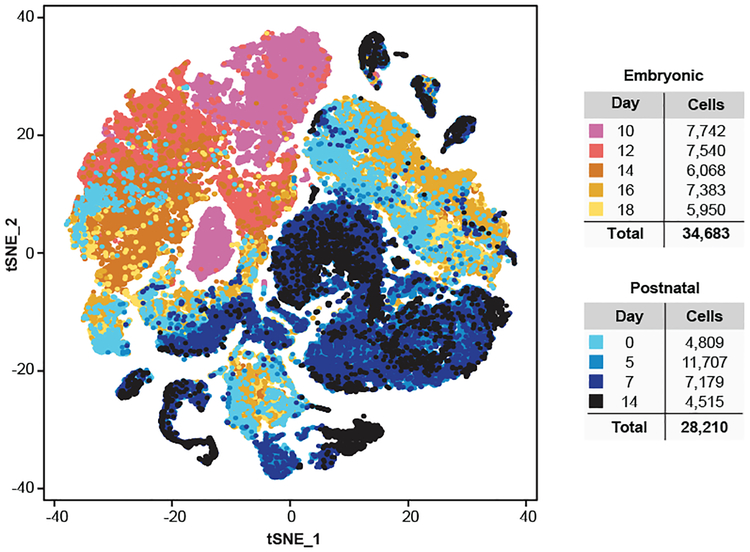
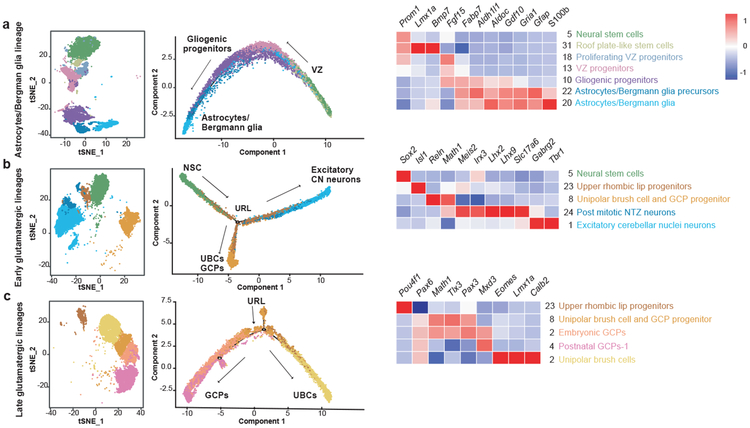
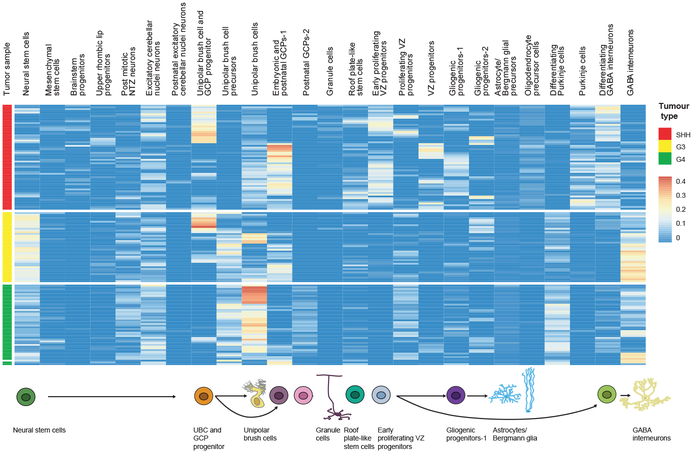
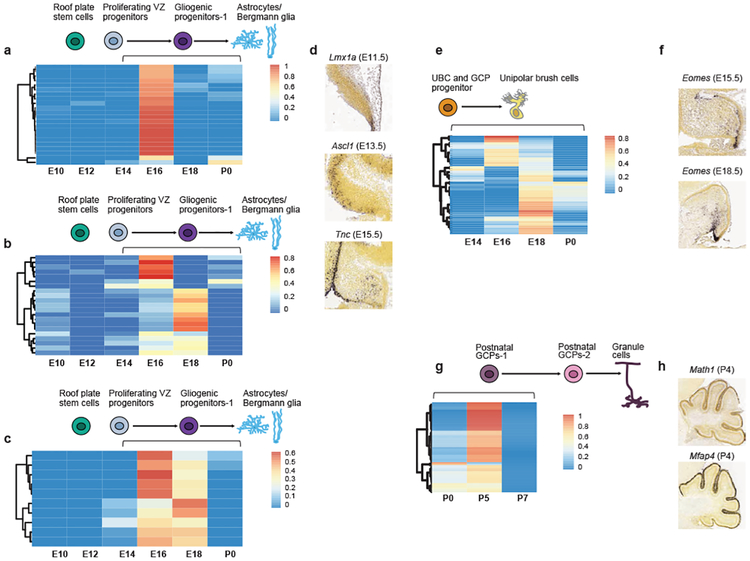
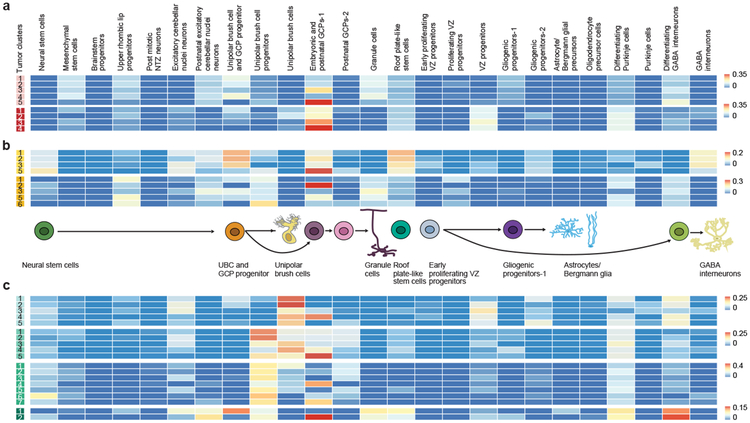
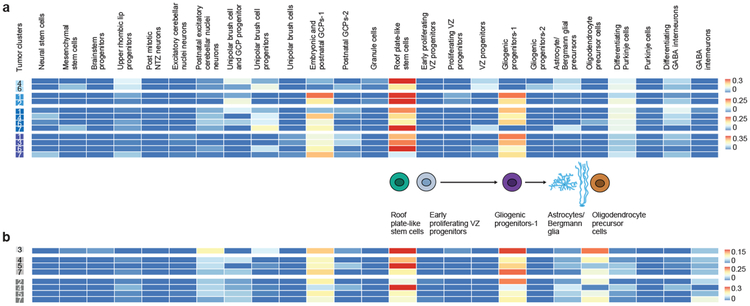
Study of the origin and development of cerebellar tumours has been hampered by the complexity and heterogeneity of cerebellar cells that change over the course of development. Here we use single-cell transcriptomics to study more than 60,000 cells from the developing mouse cerebellum and show that different molecular subgroups of childhood cerebellar tumours mirror the transcription of cells from distinct, temporally restricted cerebellar lineages. The Sonic Hedgehog medulloblastoma subgroup transcriptionally mirrors the granule cell hierarchy as expected, while group 3 medulloblastoma resembles Nestin+ stem cells, group 4 medulloblastoma resembles unipolar brush cells, and PFA/PFB ependymoma and cerebellar pilocytic astrocytoma resemble the prenatal gliogenic progenitor cells. Furthermore, single-cell transcriptomics of human childhood cerebellar tumours demonstrates that many bulk tumours contain a mixed population of cells with divergent differentiation. Our data highlight cerebellar tumours as a disorder of early brain development and provide a proximate explanation for the peak incidence of cerebellar tumours in early childhood.
Conflict of interest statement
Figures
Comment in
-
Pediatric cerebellar tumors: transcriptionally distinct but developmentally heterogeneous.Transl Cancer Res. 2020 Mar;9(3):1322-1325. doi: 10.21037/tcr.2020.01.31. Transl Cancer Res. 2020. PMID: 35117480 Free PMC article. No abstract available.
References
Methods References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
