

A vitamin-C-derived DNA modification catalysed by an algal TET homologue
- PMID: 31043749
- PMCID: PMC6628258
- DOI: 10.1038/s41586-019-1160-0
A vitamin-C-derived DNA modification catalysed by an algal TET homologue
Abstract
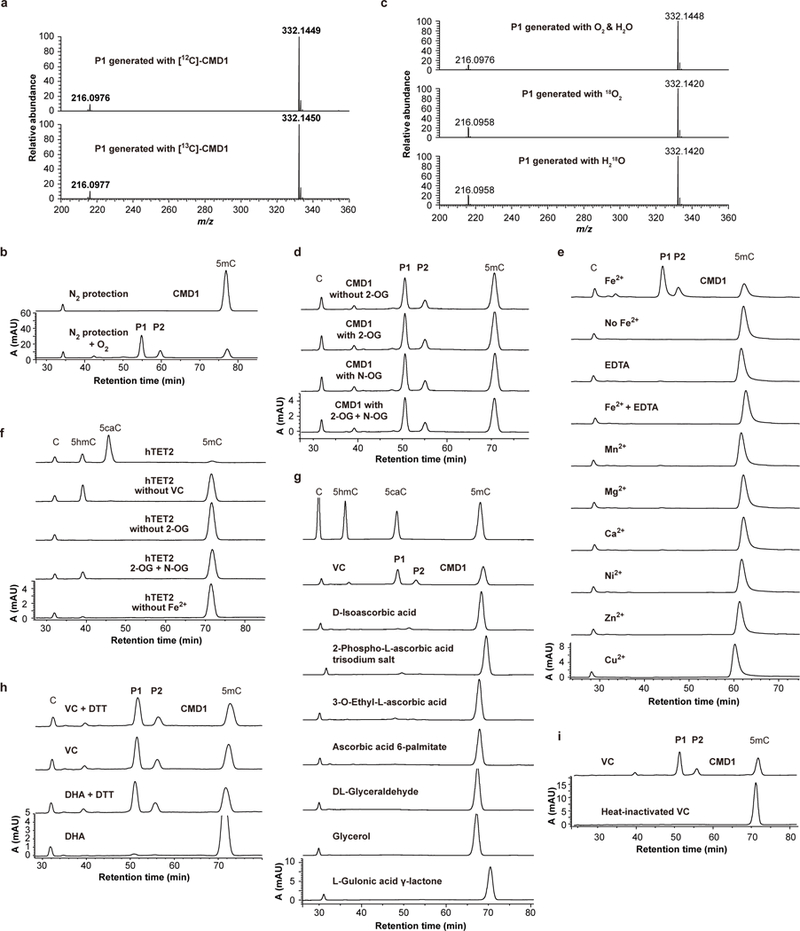
Methylation of cytosine to 5-methylcytosine (5mC) is a prevalent DNA modification found in many organisms. Sequential oxidation of 5mC by ten-eleven translocation (TET) dioxygenases results in a cascade of additional epigenetic marks and promotes demethylation of DNA in mammals1,2. However, the enzymatic activity and function of TET homologues in other eukaryotes remains largely unexplored. Here we show that the green alga Chlamydomonas reinhardtii contains a 5mC-modifying enzyme (CMD1) that is a TET homologue and catalyses the conjugation of a glyceryl moiety to the methyl group of 5mC through a carbon-carbon bond, resulting in two stereoisomeric nucleobase products. The catalytic activity of CMD1 requires Fe(II) and the integrity of its binding motif His-X-Asp, which is conserved in Fe-dependent dioxygenases3. However, unlike previously described TET enzymes, which use 2-oxoglutarate as a co-substrate4, CMD1 uses L-ascorbic acid (vitamin C) as an essential co-substrate. Vitamin C donates the glyceryl moiety to 5mC with concurrent formation of glyoxylic acid and CO2. The vitamin-C-derived DNA modification is present in the genome of wild-type C. reinhardtii but at a substantially lower level in a CMD1 mutant strain. The fitness of CMD1 mutant cells during exposure to high light levels is reduced. LHCSR3, a gene that is critical for the protection of C. reinhardtii from photo-oxidative damage under high light conditions, is hypermethylated and downregulated in CMD1 mutant cells compared to wild-type cells, causing a reduced capacity for photoprotective non-photochemical quenching. Our study thus identifies a eukaryotic DNA base modification that is catalysed by a divergent TET homologue and unexpectedly derived from vitamin C, and describes its role as a potential epigenetic mark that may counteract DNA methylation in the regulation of photosynthesis.
Figures














Comment in
-
A Novel Epigenetic Mark Derived from Vitamin C.Biochemistry. 2020 Jan 14;59(1):8-9. doi: 10.1021/acs.biochem.9b00779. Epub 2019 Sep 20. Biochemistry. 2020. PMID: 31538774 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
