

The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells
- PMID: 31048320
- PMCID: PMC6606338
- DOI: 10.1158/2159-8290.CD-19-0125
The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells
Abstract
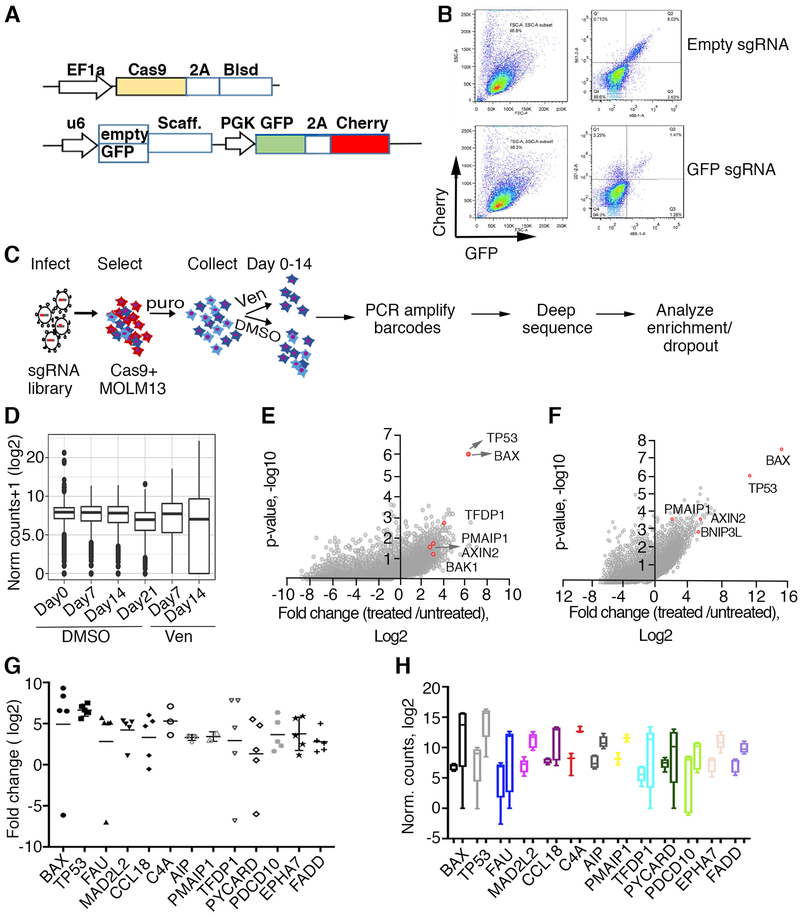
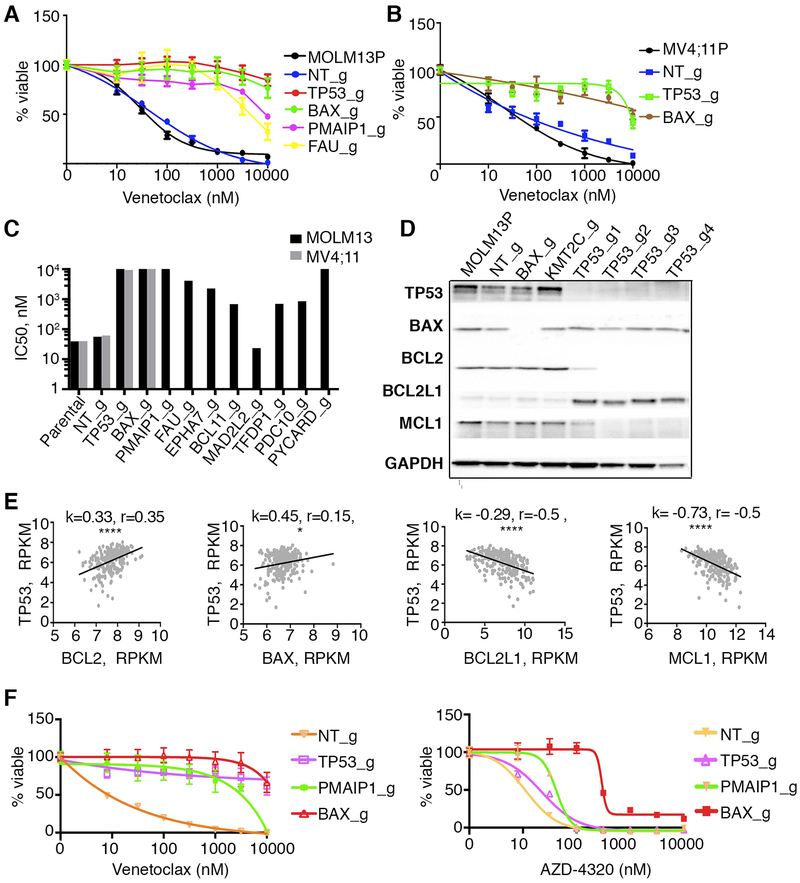
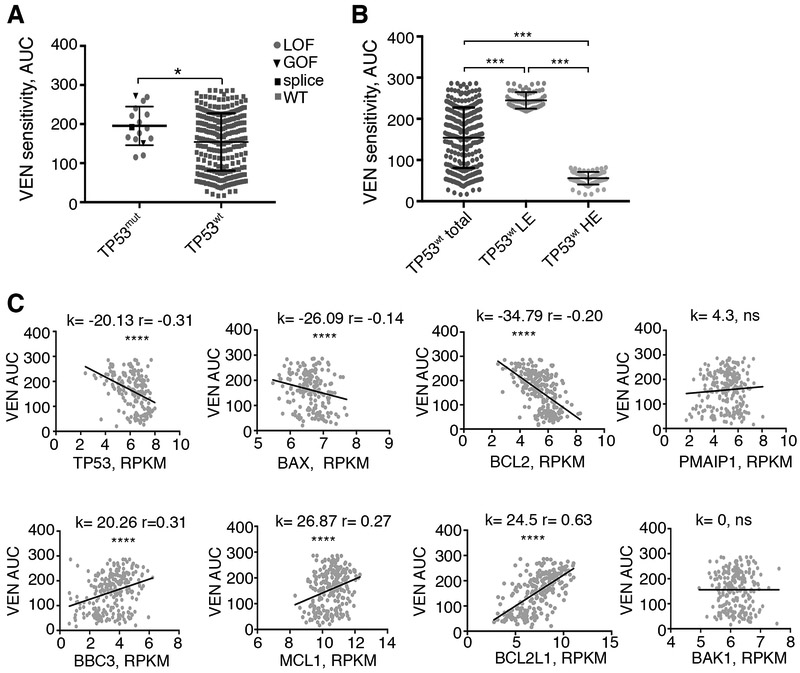
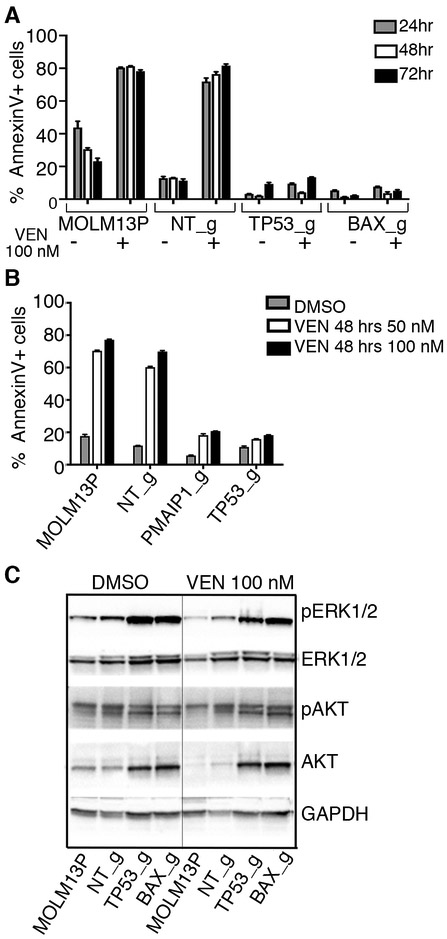
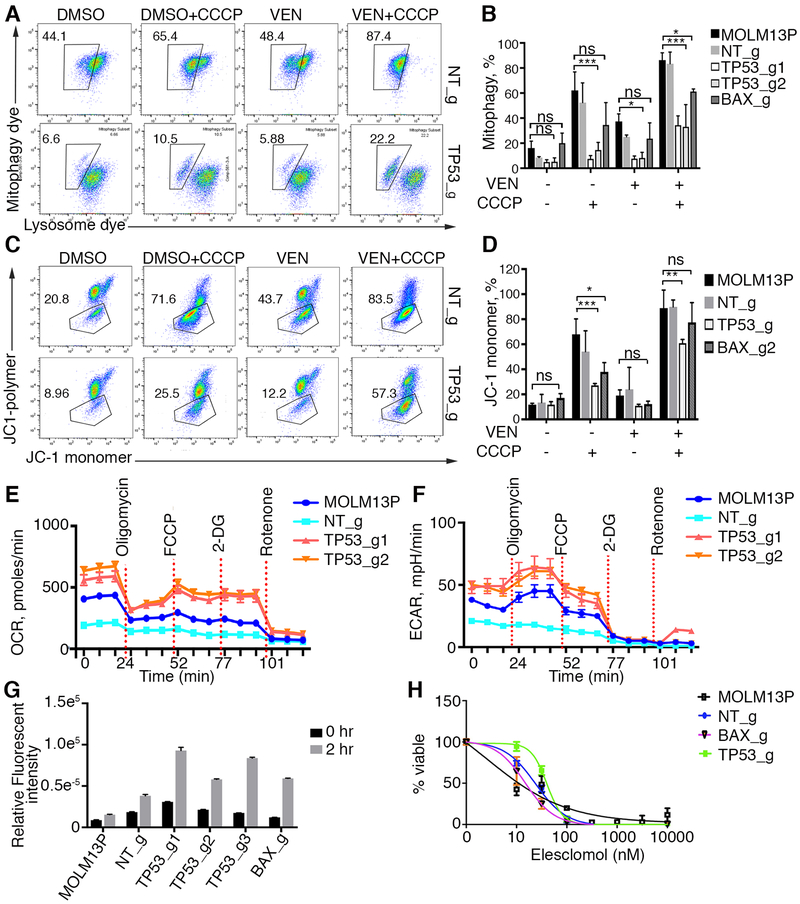
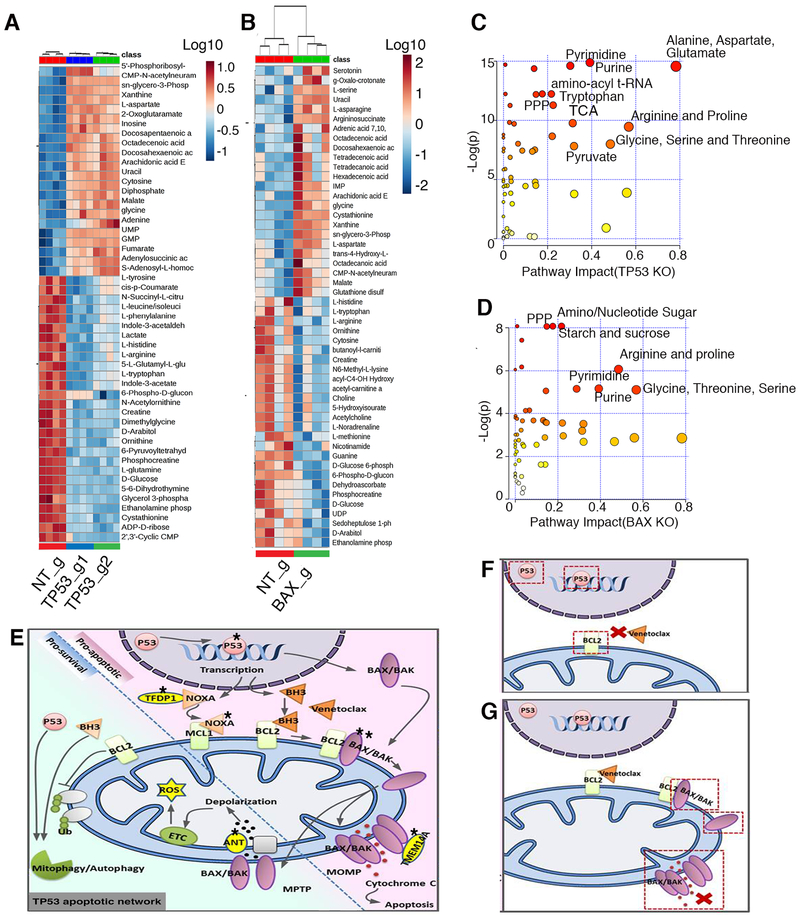
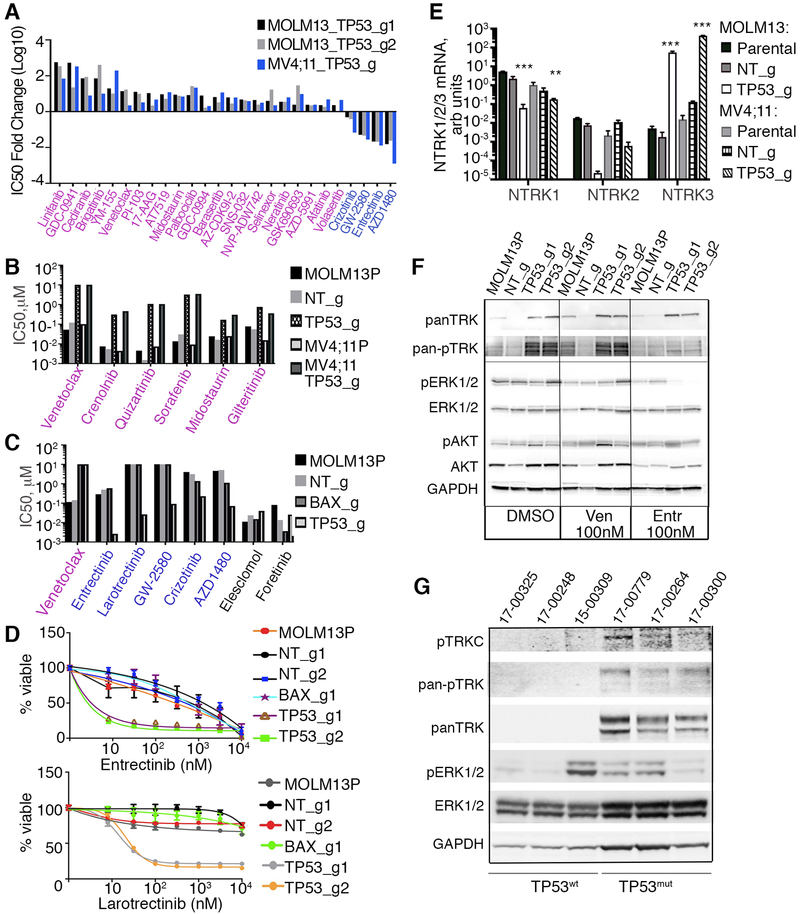
To study mechanisms underlying resistance to the BCL2 inhibitor venetoclax in acute myeloid leukemia (AML), we used a genome-wide CRISPR/Cas9 screen to identify gene knockouts resulting in drug resistance. We validated TP53, BAX, and PMAIP1 as genes whose inactivation results in venetoclax resistance in AML cell lines. Resistance to venetoclax resulted from an inability to execute apoptosis driven by BAX loss, decreased expression of BCL2, and/or reliance on alternative BCL2 family members such as BCL2L1. The resistance was accompanied by changes in mitochondrial homeostasis and cellular metabolism. Evaluation of TP53 knockout cells for sensitivities to a panel of small-molecule inhibitors revealed a gain of sensitivity to TRK inhibitors. We relate these observations to patient drug responses and gene expression in the Beat AML dataset. Our results implicate TP53, the apoptotic network, and mitochondrial functionality as drivers of venetoclax response in AML and suggest strategies to overcome resistance. SIGNIFICANCE: AML is challenging to treat due to its heterogeneity, and single-agent therapies have universally failed, prompting a need for innovative drug combinations. We used a genetic approach to identify genes whose inactivation contributes to drug resistance as a means of forming preferred drug combinations to improve AML treatment.See related commentary by Savona and Rathmell, p. 831.This article is highlighted in the In This Issue feature, p. 813.
©2019 American Association for Cancer Research.
Figures
References
-
- Hanahan D and Weinberg RA, Hallmarks of cancer: the next generation. Cell, 2011. 144(5): p. 646–74. - PubMed
-
- Letai AG, Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer, 2008. 8(2): p. 121–32. - PubMed
-
- Khoo KH, Verma CS, and Lane DP, Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov, 2014. 13(3): p. 217–36. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
