

Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment
- PMID: 31048321
- PMCID: PMC6606342
- DOI: 10.1158/2159-8290.CD-19-0117
Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment
Abstract
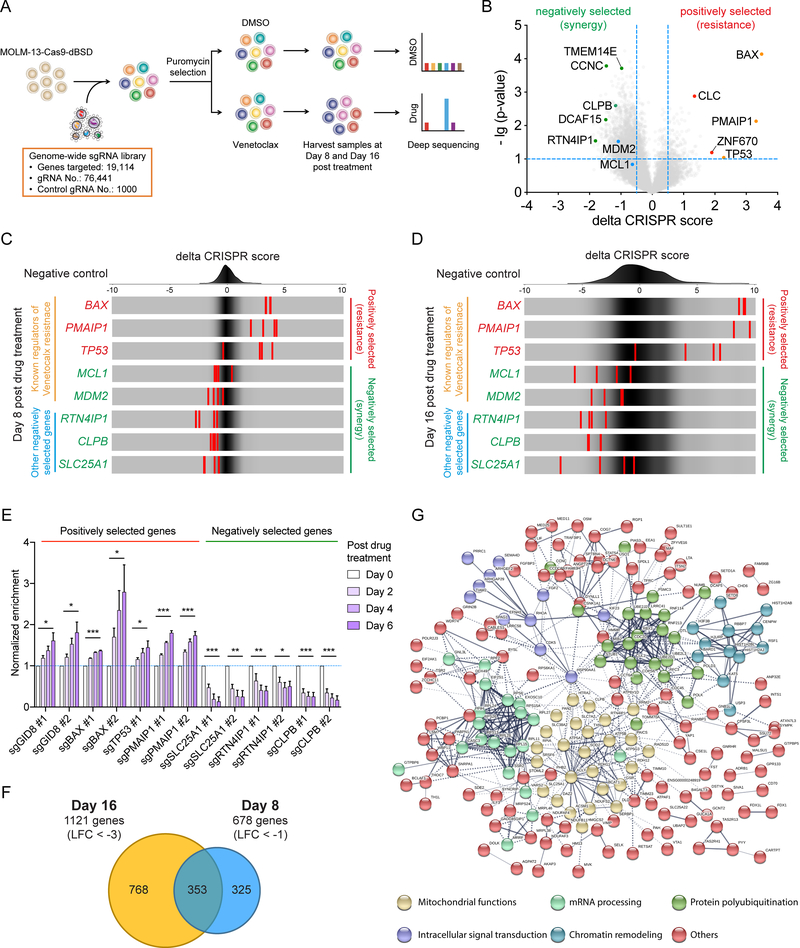
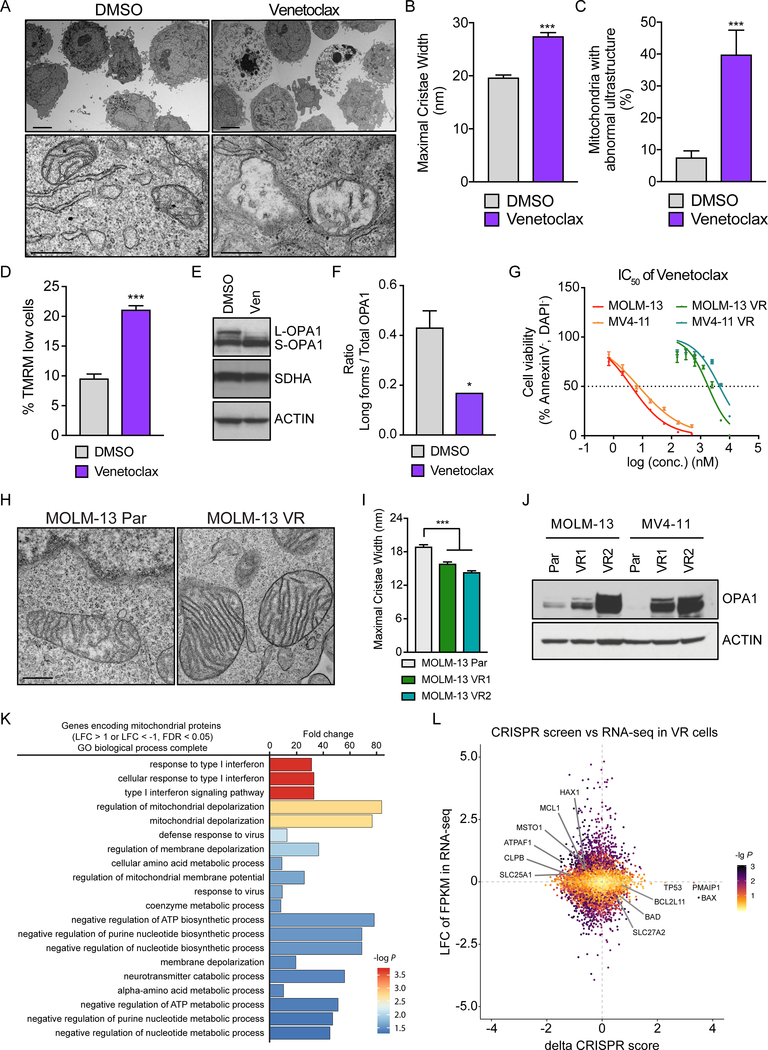
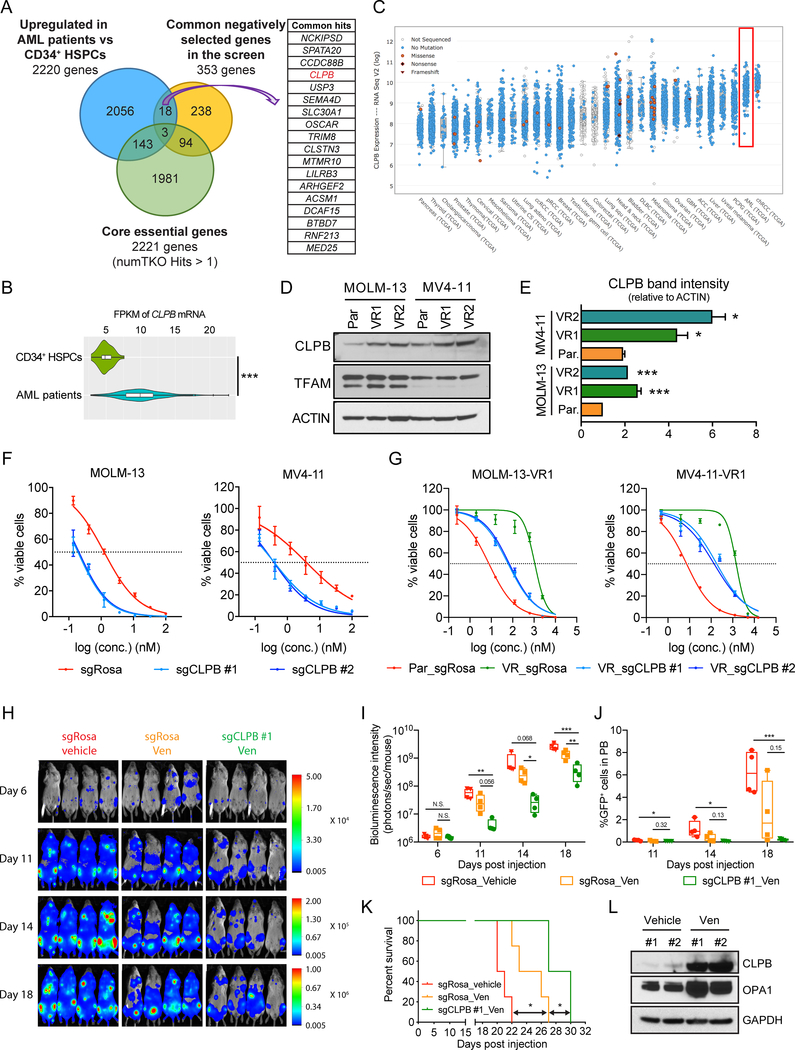
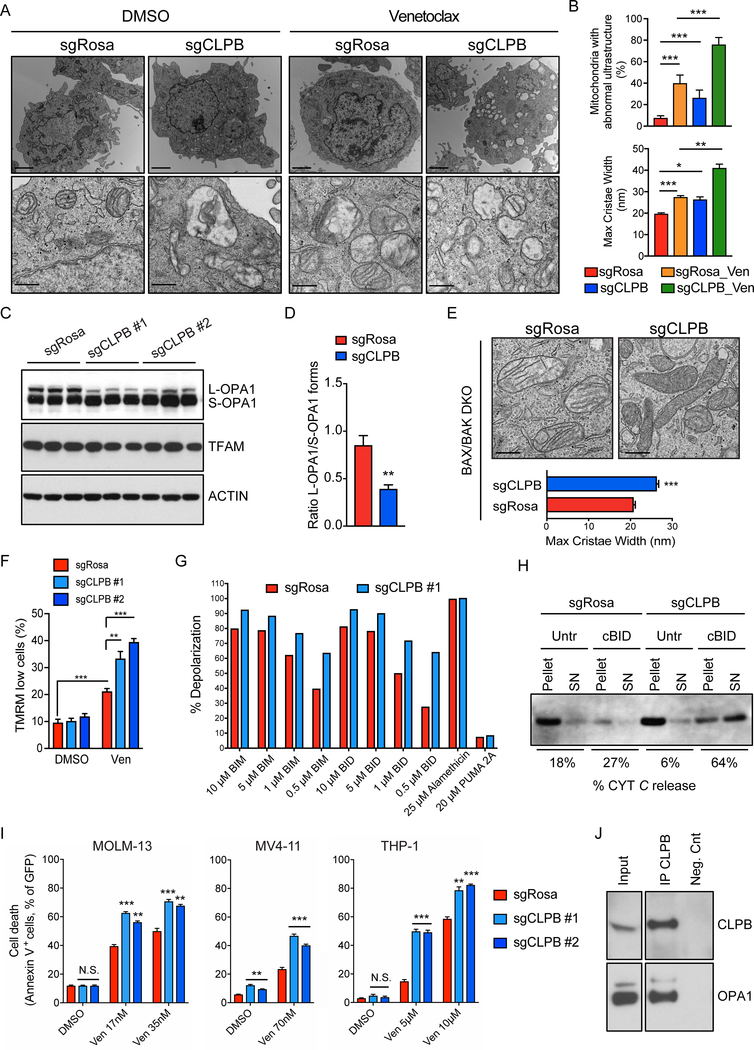
The BCL2 family plays important roles in acute myeloid leukemia (AML). Venetoclax, a selective BCL2 inhibitor, has received FDA approval for the treatment of AML. However, drug resistance ensues after prolonged treatment, highlighting the need for a greater understanding of the underlying mechanisms. Using a genome-wide CRISPR/Cas9 screen in human AML, we identified genes whose inactivation sensitizes AML blasts to venetoclax. Genes involved in mitochondrial organization and function were significantly depleted throughout our screen, including the mitochondrial chaperonin CLPB. We demonstrated that CLPB is upregulated in human AML, it is further induced upon acquisition of venetoclax resistance, and its ablation sensitizes AML to venetoclax. Mechanistically, CLPB maintains the mitochondrial cristae structure via its interaction with the cristae-shaping protein OPA1, whereas its loss promotes apoptosis by inducing cristae remodeling and mitochondrial stress responses. Overall, our data suggest that targeting mitochondrial architecture may provide a promising approach to circumvent venetoclax resistance. SIGNIFICANCE: A genome-wide CRISPR/Cas9 screen reveals genes involved in mitochondrial biological processes participate in the acquisition of venetoclax resistance. Loss of the mitochondrial protein CLPB leads to structural and functional defects of mitochondria, hence sensitizing AML cells to apoptosis. Targeting CLPB synergizes with venetoclax and the venetoclax/azacitidine combination in AML in a p53-independent manner.See related commentary by Savona and Rathmell, p. 831.This article is highlighted in the In This Issue feature, p. 813.
©2019 American Association for Cancer Research.
Conflict of interest statement
Conflicts of interest
R.T. was an advisory board member for AbbVie. No potential conflicts of interest are disclosed by other authors.
Figures
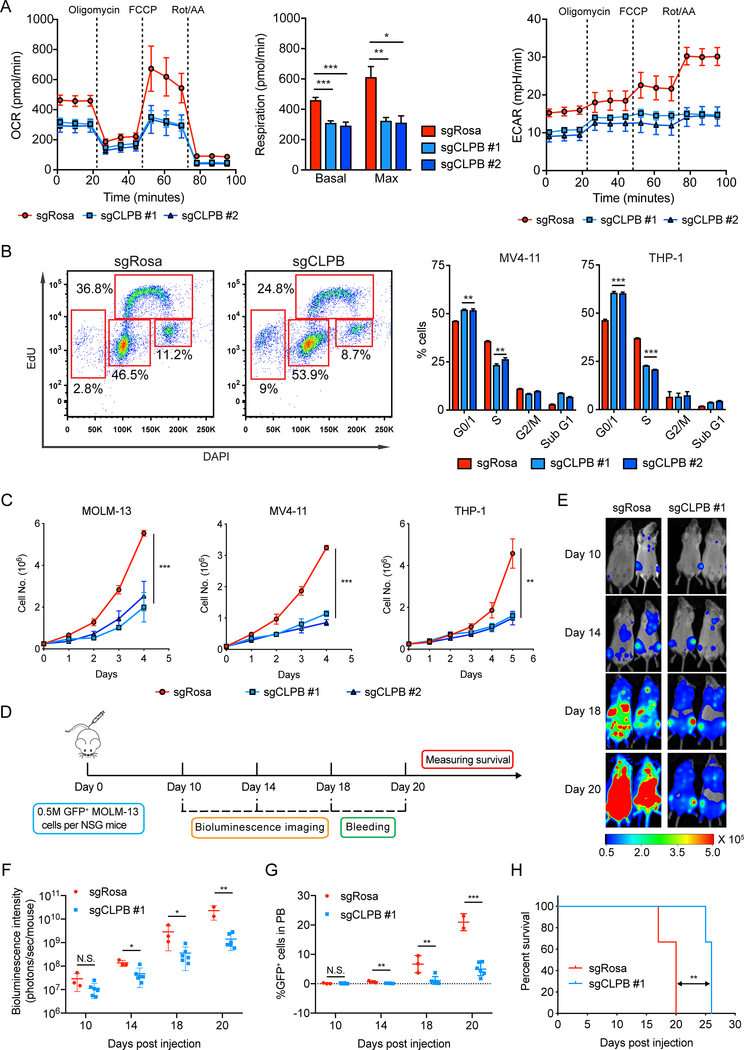
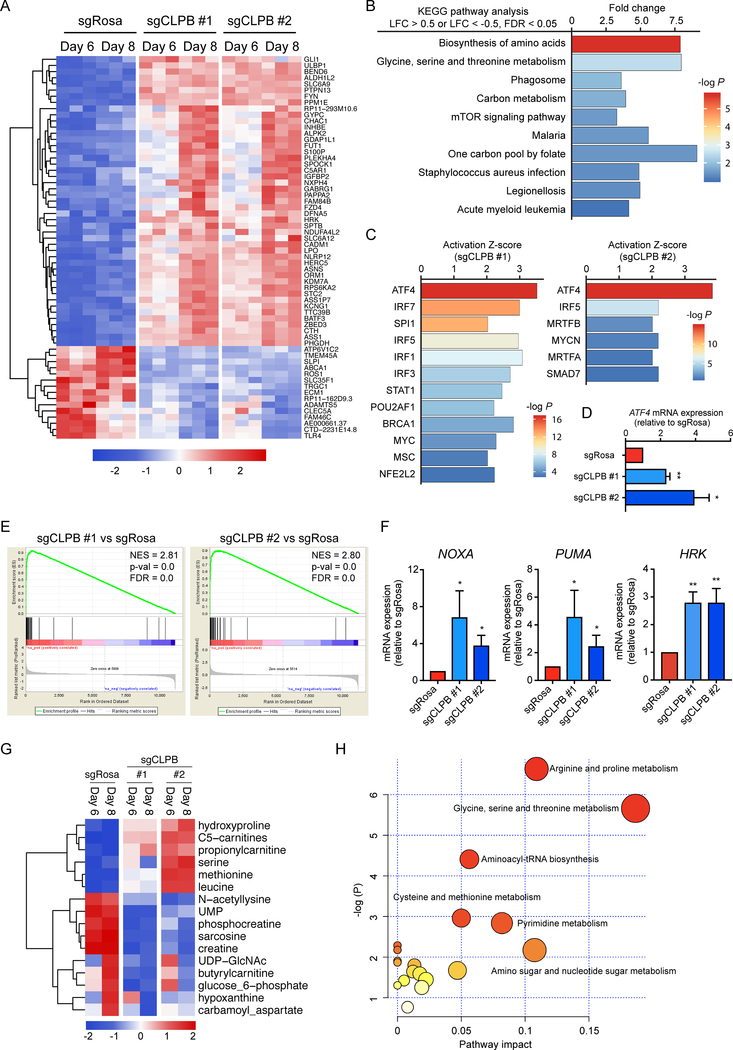
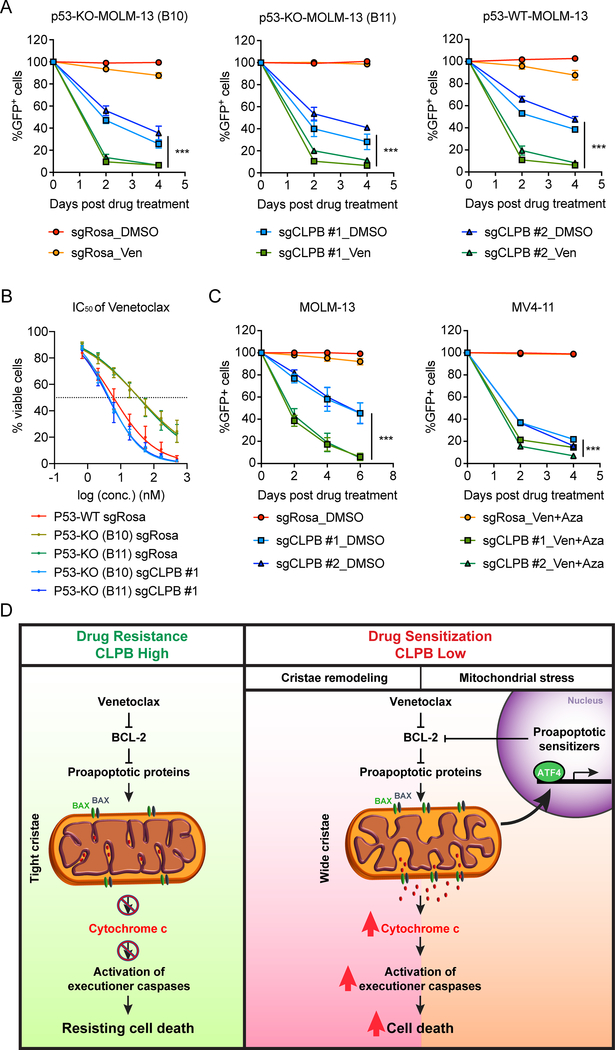
Comment in
-
Mitochondrial Homeostasis in AML and Gasping for Response in Resistance to BCL2 Blockade.Cancer Discov. 2019 Jul;9(7):831-833. doi: 10.1158/2159-8290.CD-19-0510. Cancer Discov. 2019. PMID: 31262744 Free PMC article.
References
-
- Campos L, Rouault JP, Sabido O, Oriol P, Roubi N, Vasselon C, et al. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993;81:3091–6. - PubMed
-
- Chao DT and Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. - PubMed
-
- Antignani A and Youle RJ. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr Opin Cell Biol. 2006;18:685–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA173636/CA/NCI NIH HHS/United States
- R01 CA133379/CA/NCI NIH HHS/United States
- R01 CA202027/CA/NCI NIH HHS/United States
- R01 CA228135/CA/NCI NIH HHS/United States
- R01 CA178979/CA/NCI NIH HHS/United States
- R01 CA178394/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 CA216421/CA/NCI NIH HHS/United States
- T32 GM007491/GM/NIGMS NIH HHS/United States
- R01 CA169784/CA/NCI NIH HHS/United States
- S10 RR023704/RR/NCRR NIH HHS/United States
- P30 CA016087/CA/NCI NIH HHS/United States
- S10 OD010582/OD/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
