

Targeting RIPK1 for the treatment of human diseases
- PMID: 31048504
- PMCID: PMC6525537
- DOI: 10.1073/pnas.1901179116
Targeting RIPK1 for the treatment of human diseases
Abstract
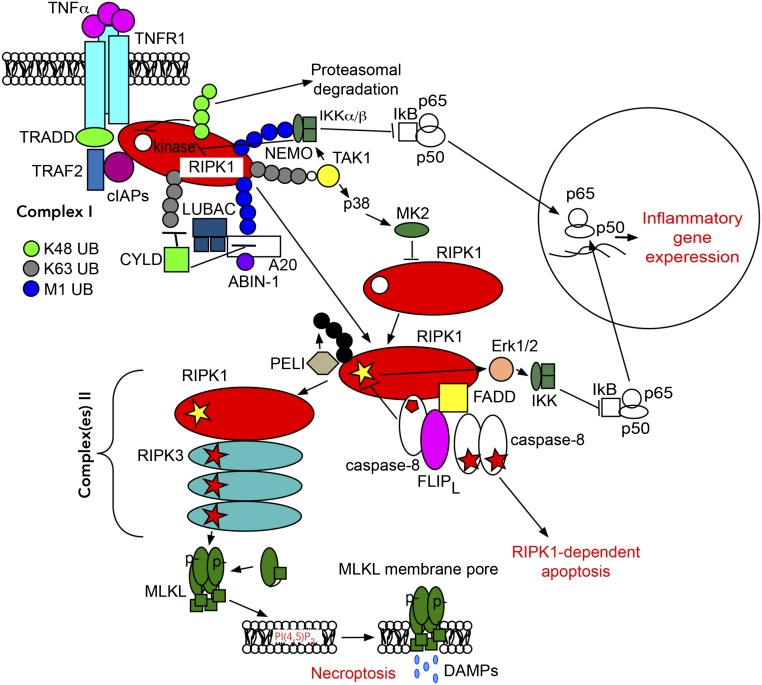
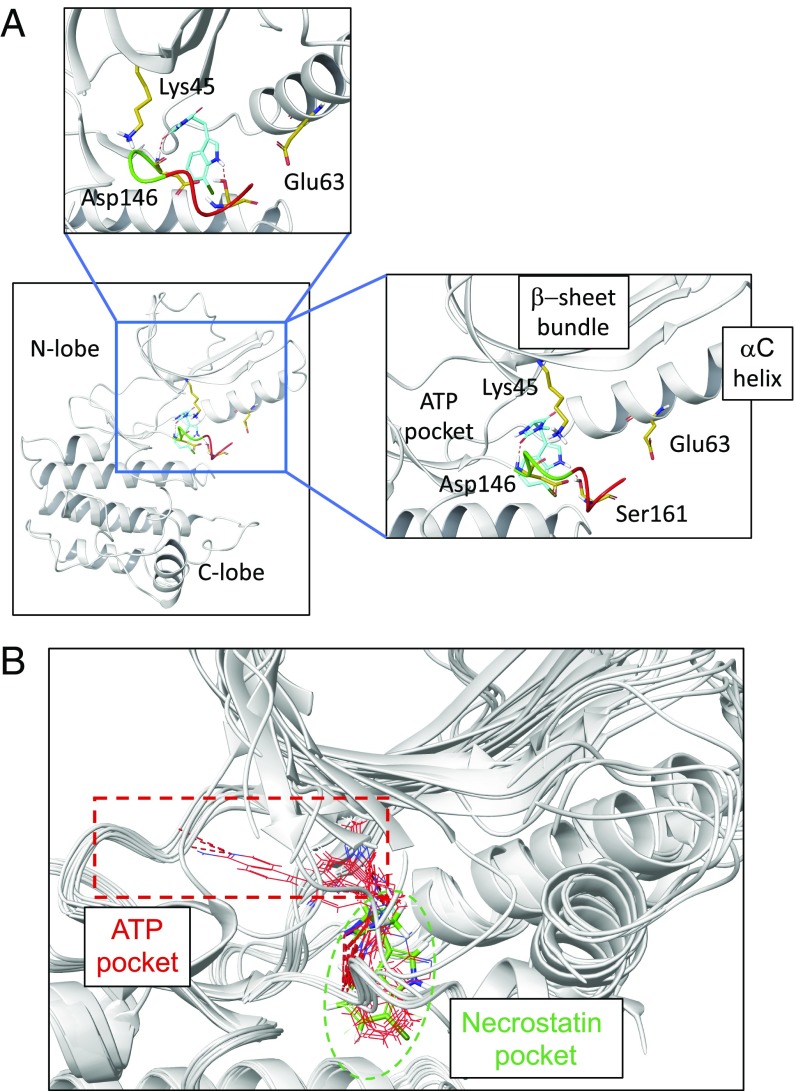
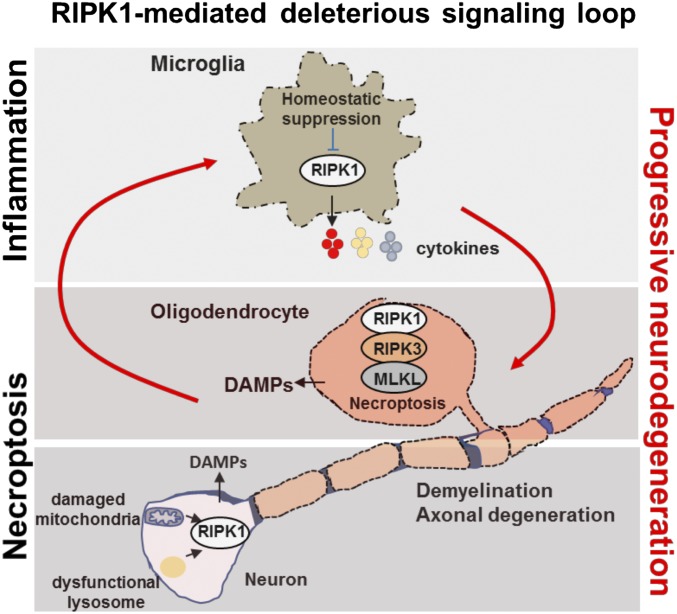
RIPK1 kinase has emerged as a promising therapeutic target for the treatment of a wide range of human neurodegenerative, autoimmune, and inflammatory diseases. This was supported by extensive studies which demonstrated that RIPK1 is a key mediator of apoptotic and necrotic cell death as well as inflammatory pathways. Furthermore, human genetic evidence has linked the dysregulation of RIPK1 to the pathogenesis of ALS as well as other inflammatory and neurodegenerative diseases. Importantly, unique allosteric small-molecule inhibitors of RIPK1 that offer high selectivity have been developed. These molecules can penetrate the blood-brain barrier, thus offering the possibility to target neuroinflammation and cell death which drive various neurologic conditions including Alzheimer's disease, ALS, and multiple sclerosis as well as acute neurological diseases such as stroke and traumatic brain injuries. We discuss the current understanding of RIPK1 regulatory mechanisms and emerging evidence for the pathological roles of RIPK1 in human diseases, especially in the context of the central nervous systems.
Keywords: RIPK1; apoptosis; inflammation; necroptosis; neurodegeneration.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
Conflict of interest statement: J.Y. is a consultant for Denali Therapeutics. D.O. is an employee of Sanofi.
Figures
References
-
- Degterev A, Boyce M, Yuan J (2003) A decade of caspases. Oncogene 22:8543–8567. - PubMed
-
- Callus BA, Vaux DL (2007) Caspase inhibitors: Viral, cellular and chemical. Cell Death Differ 14:73–78. - PubMed
-
- Wallach D, Kang TB, Dillon CP, Green DR (2016) Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 352:aaf2154. - PubMed
-
- Zheng TS, Flavell RA (2000) Divinations and surprises: Genetic analysis of caspase function in mice. Exp Cell Res 256:67–73. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
