

In silico structural elucidation of RNA-dependent RNA polymerase towards the identification of potential Crimean-Congo Hemorrhagic Fever Virus inhibitors
- PMID: 31048746
- PMCID: PMC6497722
- DOI: 10.1038/s41598-019-43129-2
In silico structural elucidation of RNA-dependent RNA polymerase towards the identification of potential Crimean-Congo Hemorrhagic Fever Virus inhibitors
Abstract
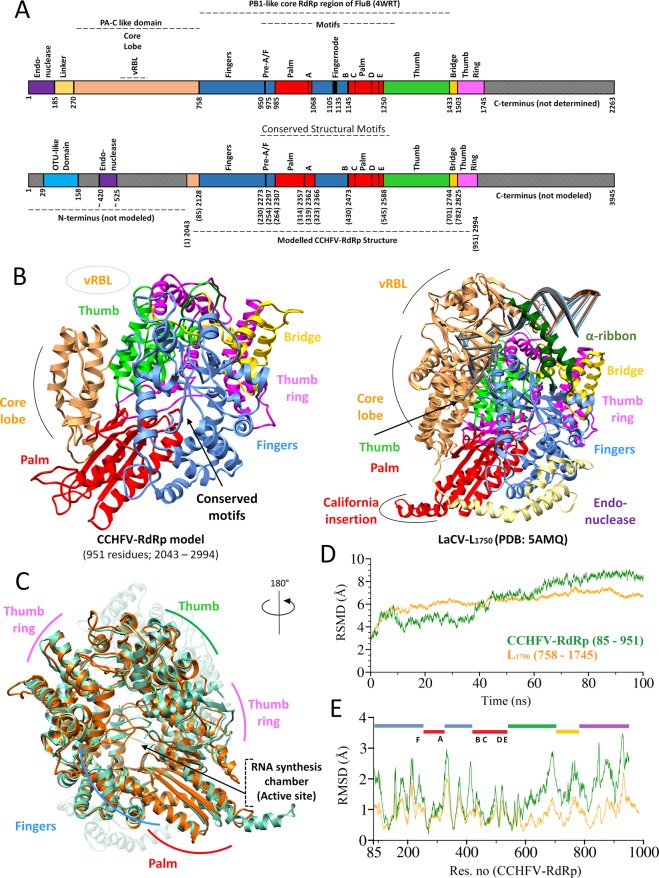
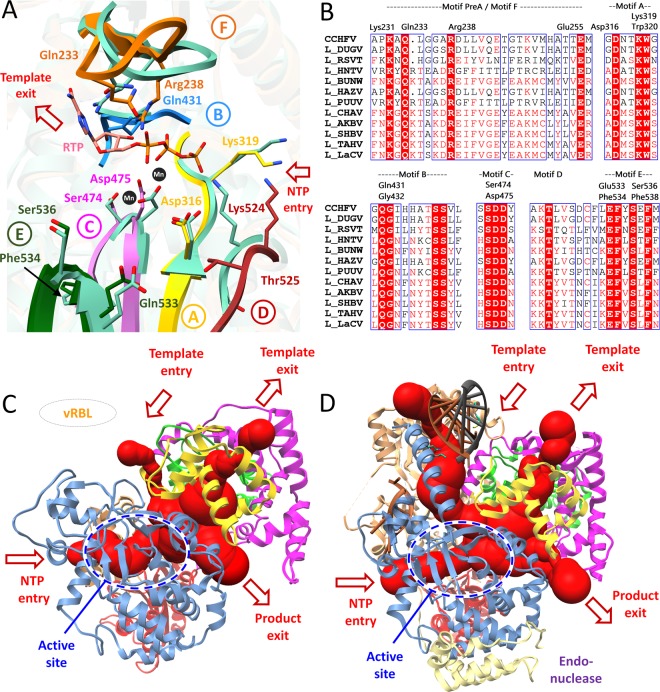
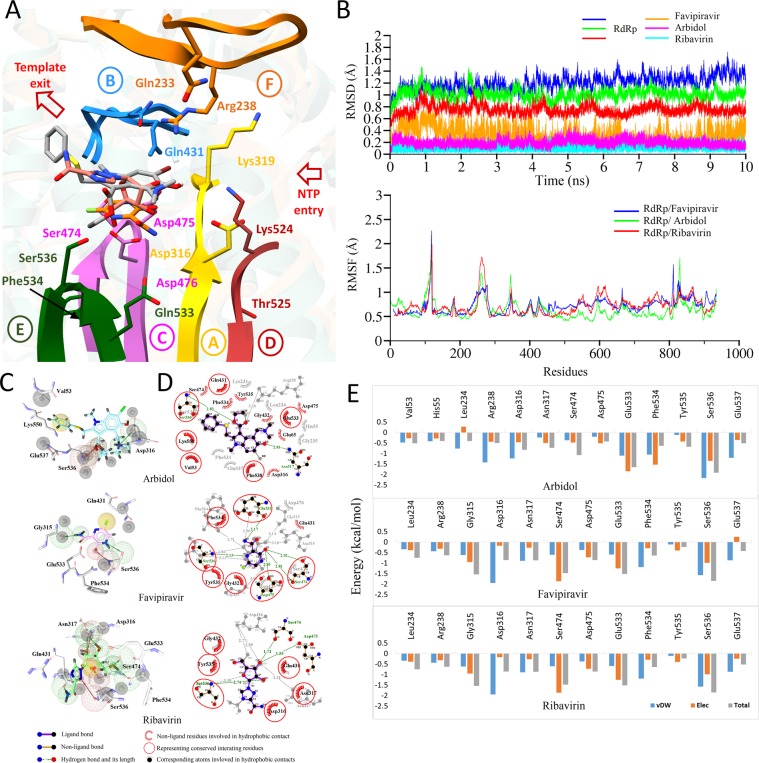
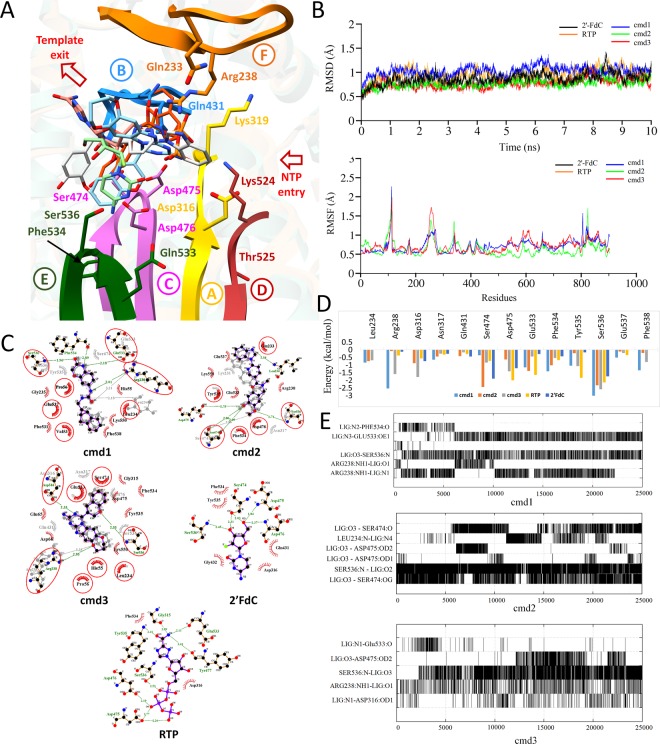
The Crimean-Congo Hemorrhagic Fever virus (CCHFV) is a segmented negative single-stranded RNA virus (-ssRNA) which causes severe hemorrhagic fever in humans with a mortality rate of ~50%. To date, no vaccine has been approved. Treatment is limited to supportive care with few investigational drugs in practice. Previous studies have identified viral RNA dependent RNA Polymerase (RdRp) as a potential drug target due to its significant role in viral replication and transcription. Since no crystal structure is available yet, we report the structural elucidation of CCHFV-RdRp by in-depth homology modeling. Even with low sequence identity, the generated model suggests a similar overall structure as previously reported RdRps. More specifically, the model suggests the presence of structural/functional conserved RdRp motifs for polymerase function, the configuration of uniform spatial arrangement of core RdRp sub-domains, and predicted positively charged entry/exit tunnels, as seen in sNSV polymerases. Extensive pharmacophore modeling based on per-residue energy contribution with investigational drugs allowed the concise mapping of pharmacophoric features and identified potential hits. The combination of pharmacophoric features with interaction energy analysis revealed functionally important residues in the conserved motifs together with in silico predicted common inhibitory binding modes with highly potent reference compounds.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Burt FJ, Spencer DC, Leman PA, Patterson B, Swanepoel R. Investigation of tick-borne viruses as pathogens of humans in South Africa and evidence of Dugbe virus infection in a patient with prolonged thrombocytopenia. Epidemiology and Infection. 1996;116:353–361. doi: 10.1017/S0950268800052687. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
