

FastqPuri: high-performance preprocessing of RNA-seq data
- PMID: 31053060
- PMCID: PMC6500068
- DOI: 10.1186/s12859-019-2799-0
FastqPuri: high-performance preprocessing of RNA-seq data
Abstract
Background: RNA sequencing (RNA-seq) has become the standard means of analyzing gene and transcript expression in high-throughput. While previously sequence alignment was a time demanding step, fast alignment methods and even more so transcript counting methods which avoid mapping and quantify gene and transcript expression by evaluating whether a read is compatible with a transcript, have led to significant speed-ups in data analysis. Now, the most time demanding step in the analysis of RNA-seq data is preprocessing the raw sequence data, such as running quality control and adapter, contamination and quality filtering before transcript or gene quantification. To do so, many researchers chain different tools, but a comprehensive, flexible and fast software that covers all preprocessing steps is currently missing.
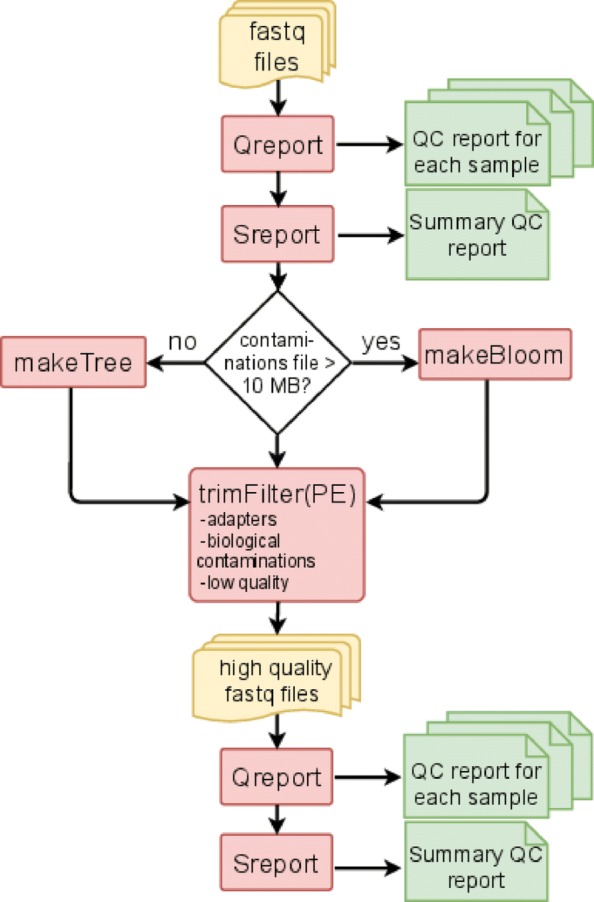
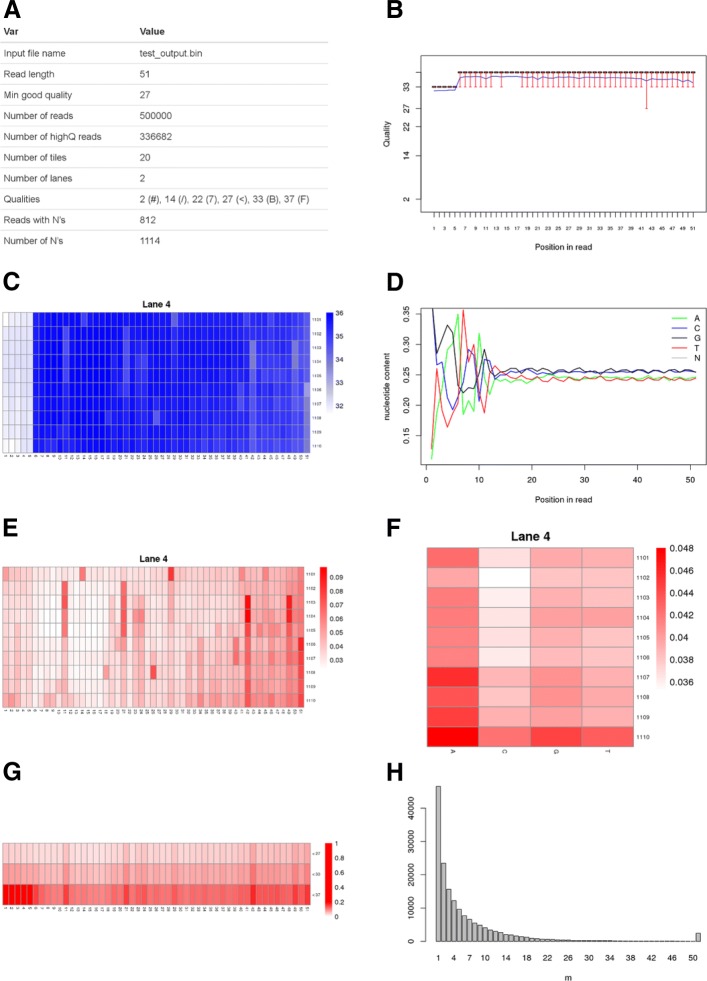
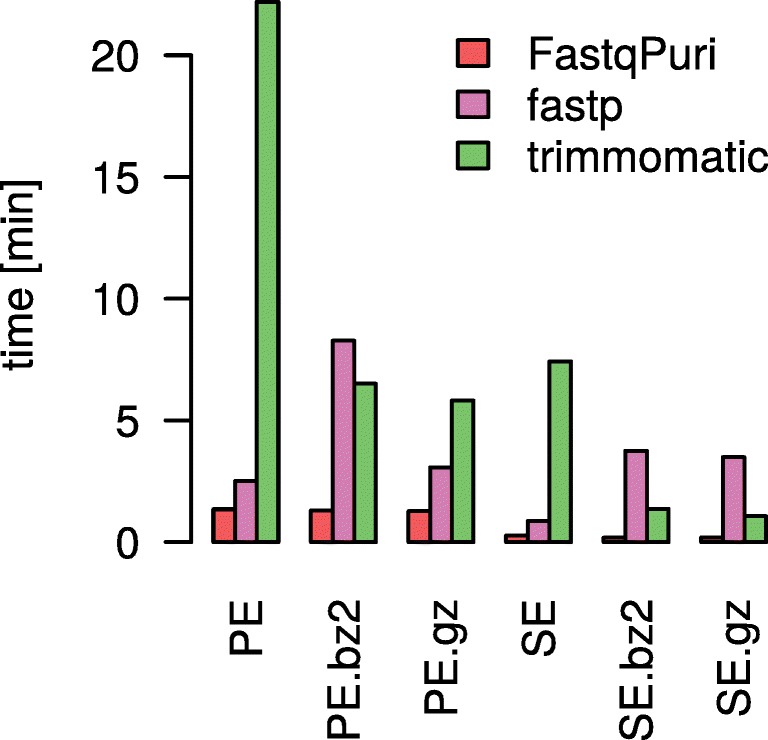
Results: We here present FastqPuri, a light-weight and highly efficient preprocessing tool for fastq data. FastqPuri provides sequence quality reports on the sample and dataset level with new plots which facilitate decision making for subsequent quality filtering. Moreover, FastqPuri efficiently removes adapter sequences and sequences from biological contamination from the data. It accepts both single- and paired-end data in uncompressed or compressed fastq files. FastqPuri can be run stand-alone and is suitable to be run within pipelines. We benchmarked FastqPuri against existing tools and found that FastqPuri is superior in terms of speed, memory usage, versatility and comprehensiveness.
Conclusions: FastqPuri is a new tool which covers all aspects of short read sequence data preprocessing. It was designed for RNA-seq data to meet the needs for fast preprocessing of fastq data to allow transcript and gene counting, but it is suitable to process any short read sequencing data of which high sequence quality is needed, such as for genome assembly or SNV (single nucleotide variant) detection. FastqPuri is most flexible in filtering undesired biological sequences by offering two approaches to optimize speed and memory usage dependent on the total size of the potential contaminating sequences. FastqPuri is available at https://github.com/jengelmann/FastqPuri . It is implemented in C and R and licensed under GPL v3.
Keywords: Preprocessing; Quality control; RNA-seq; Sequence data; fastq.
Conflict of interest statement
Ethics approval and consent to participate
Human cell sampling has been approved by the ethics committee of the University Medical Center Göttingen (Ethikkommission der Universitätsmedizin Göttingen), reference number 16/5/18An. All human participants granted written, informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures



References
-
- Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. 14.05.2018 Available online at http://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed 14 May 2018.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
