

Perspectives in genetic counseling for spinal muscular atrophy in the new therapeutic era: early pre-symptomatic intervention and test in minors
- PMID: 31053787
- PMCID: PMC6871529
- DOI: 10.1038/s41431-019-0415-4
Perspectives in genetic counseling for spinal muscular atrophy in the new therapeutic era: early pre-symptomatic intervention and test in minors
Abstract
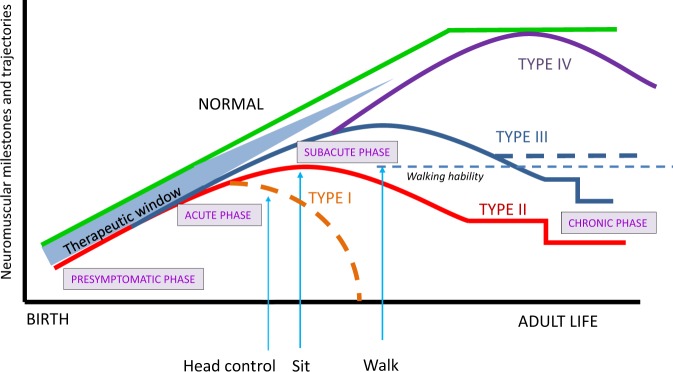
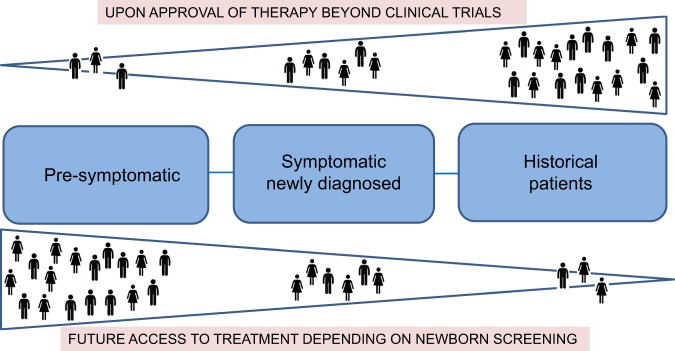
Spinal muscular atrophy (SMA) is an autosomal-recessive neuromuscular disorder representing a continuous spectrum of muscular weakness ranging from compromised neonates to adults with minimal manifestations. Patients show homozygous absence or disease-causing variants of the SMN1 gene (-/- or 0/0) and in carriers only one copy is absent or mutated (1/0). Genetic diagnosis and counseling in SMA present several challenges, including the existence of carriers (2/0) that are undistinguishable of non-carriers (1/1) with current genetic testing methods and the report of patients (0/0) with very mild manifestations and even asymptomatic that are discovered when a full symptomatic case appears in the family. Younger asymptomatic siblings of symptomatic SMA patients are usually never tested until adolescence or adult life. However, following regulatory approval of the first tailored treatment for SMA, the prospects for care of these patients have changed. Early testing, including pre-symptomatic newborn screening and confirmation of diagnosis would change proactive measures and opportunities for therapy based in the actual landscape of new treatments. This review discusses the challenges and new perspectives of genetic counseling in SMA.
Conflict of interest statement
E.F.T. has received grant support to conduct clinical trials on SMA from Ionis/Biogen and serves as a consultant to Biogen, AveXis, Roche, Biologix, and Cytokinetics. The other author declares that she has no conflict of interest.
Figures

References
-
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy- determining gene. Cell. 1995;80:155–65. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
