

De Novo DNA Methylation at Imprinted Loci during Reprogramming into Naive and Primed Pluripotency
- PMID: 31056481
- PMCID: PMC6524733
- DOI: 10.1016/j.stemcr.2019.04.008
De Novo DNA Methylation at Imprinted Loci during Reprogramming into Naive and Primed Pluripotency
Abstract
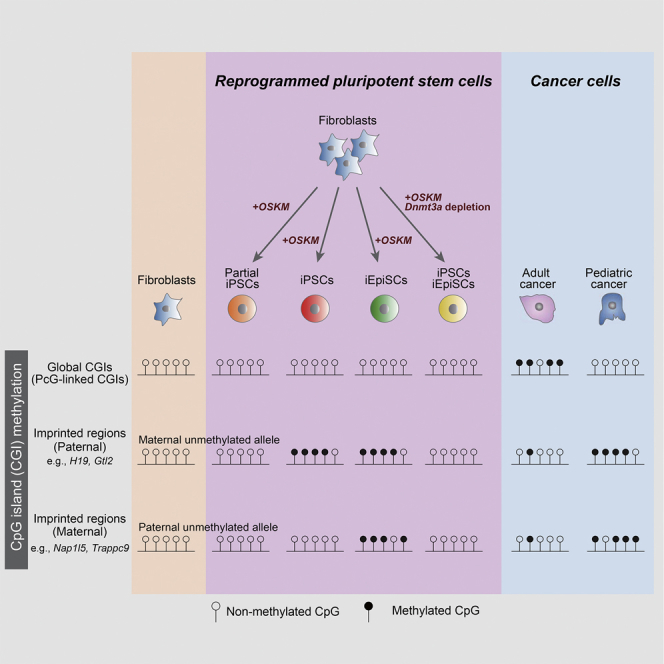
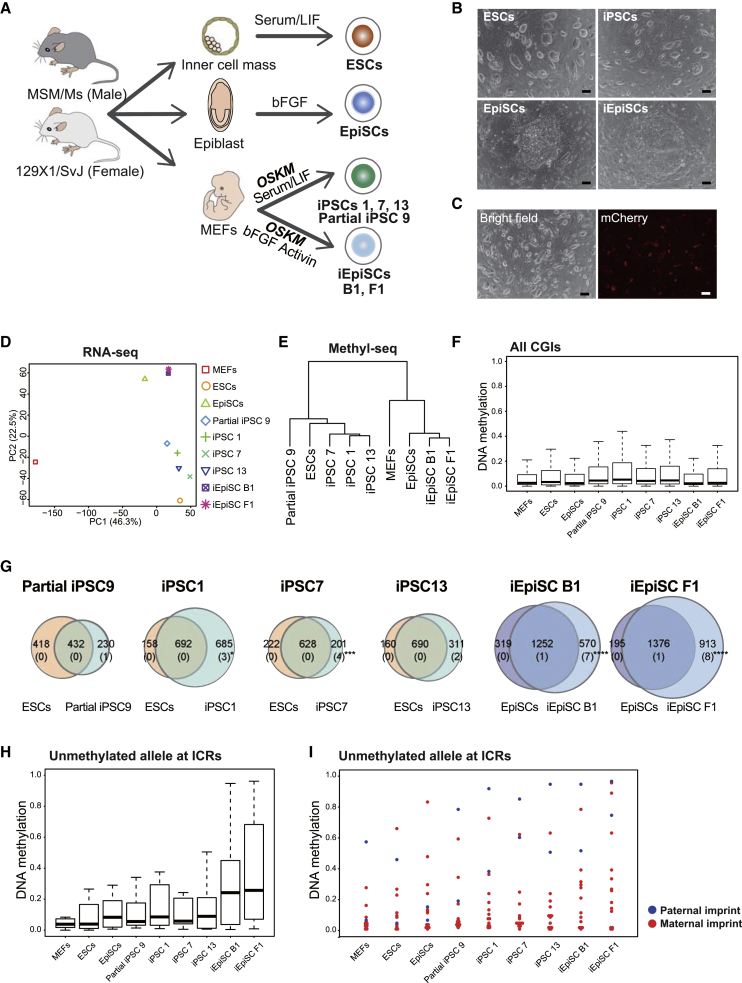
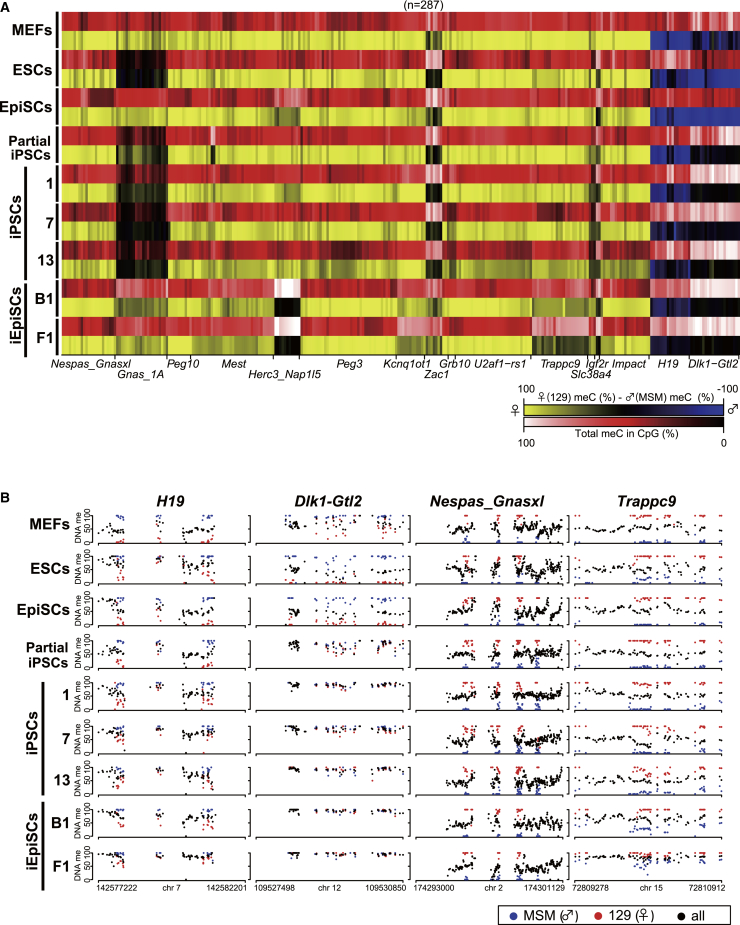
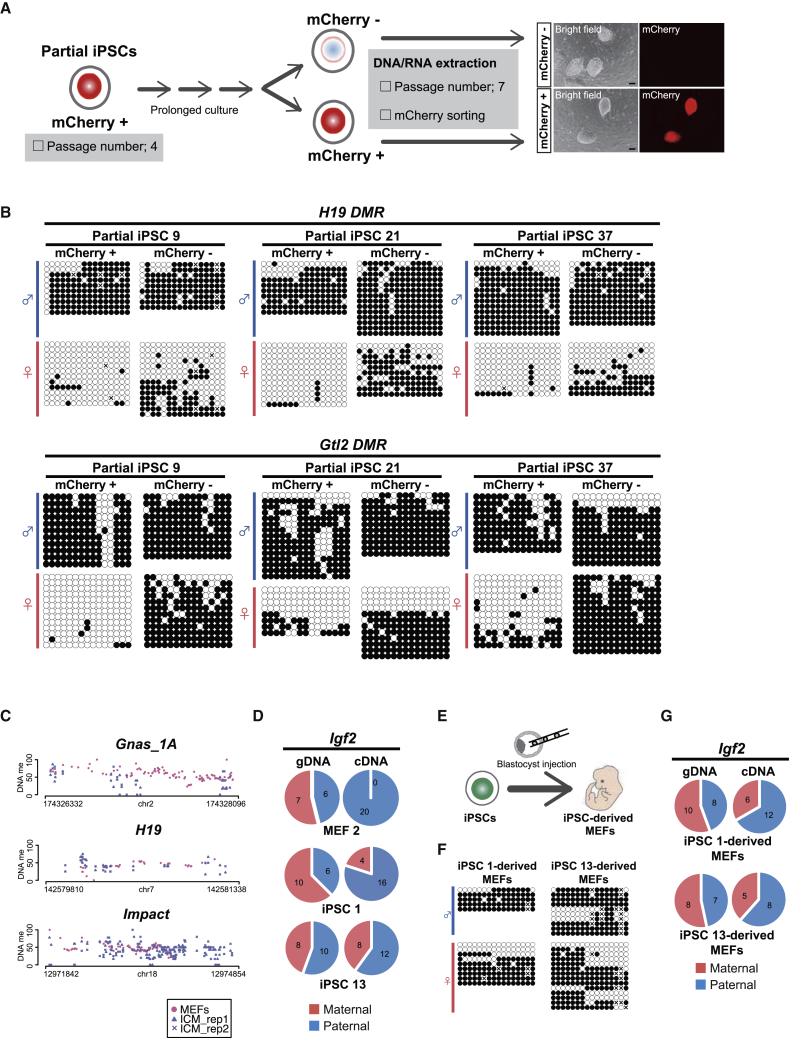
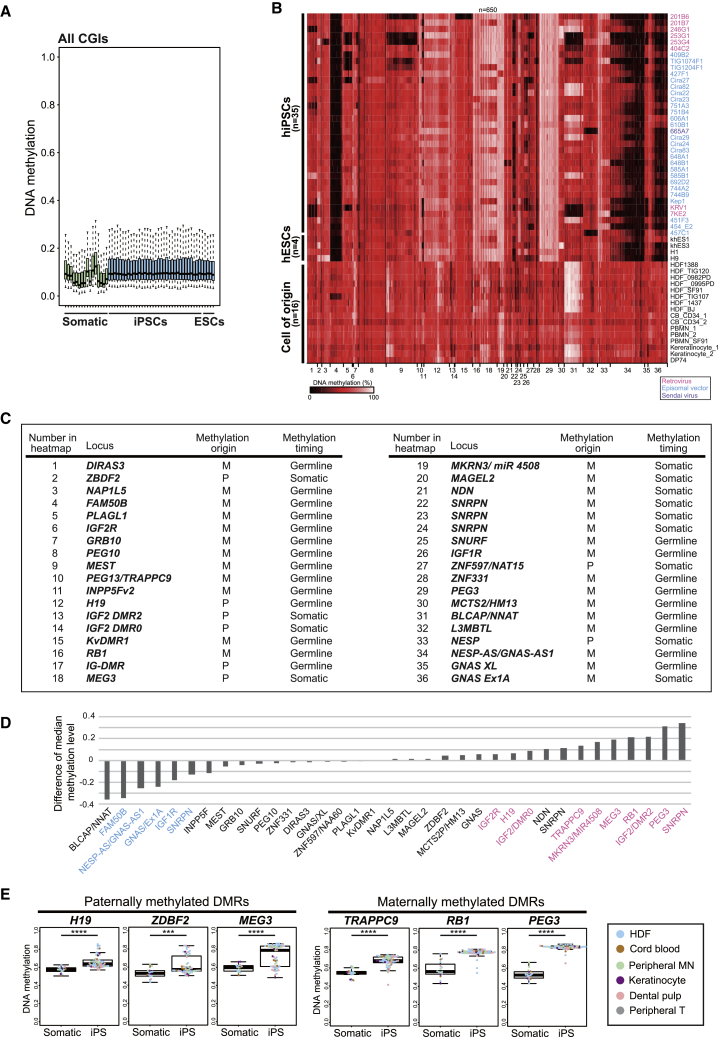
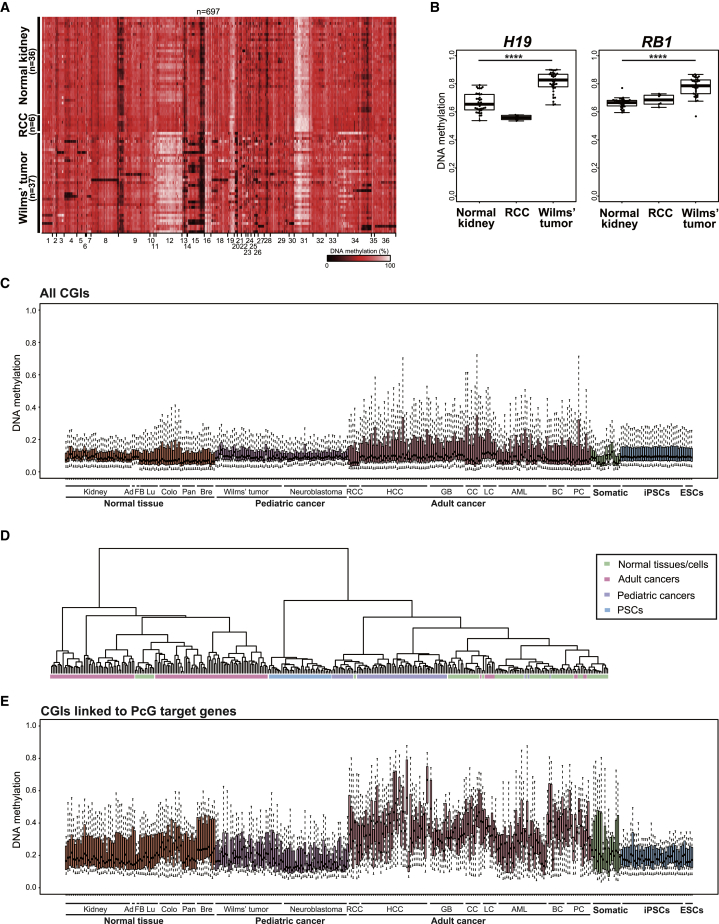
CpG islands (CGIs) including those at imprinting control regions (ICRs) are protected from de novo methylation in somatic cells. However, many cancers often exhibit CGI hypermethylation, implying that the machinery is impaired in cancer cells. Here, we conducted a comprehensive analysis of CGI methylation during somatic cell reprogramming. Although most CGIs remain hypomethylated, a small subset of CGIs, particularly at several ICRs, was often de novo methylated in reprogrammed pluripotent stem cells (PSCs). Such de novo ICR methylation was linked with the silencing of reprogramming factors, which occurs at a late stage of reprogramming. The ICR-preferred CGI hypermethylation was similarly observed in human PSCs. Mechanistically, ablation of Dnmt3a prevented PSCs from de novo ICR methylation. Notably, the ICR-preferred CGI hypermethylation was observed in pediatric cancers, while adult cancers exhibit genome-wide CGI hypermethylation. These results may have important implications in the pathogenesis of pediatric cancers and the application of PSCs.
Keywords: CpG islands; DNA methylation; Dnmt3a; genomic imprinting; naive and primed pluripotency; pediatric cancers; pluripotent stem cells; reprogramming.
Copyright © 2019 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Bar S., Schachter M., Eldar-Geva T., Benvenisty N. Large-scale analysis of loss of imprinting in human pluripotent stem cells. Cell Rep. 2017;19:957–968. - PubMed
-
- Bourc'his D., Xu G.L., Lin C.S., Bollman B., Bestor T.H. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
