

Diagnosis, Phenotype, and Molecular Genetics of Congenital Analbuminemia
- PMID: 31057599
- PMCID: PMC6478806
- DOI: 10.3389/fgene.2019.00336
Diagnosis, Phenotype, and Molecular Genetics of Congenital Analbuminemia
Abstract
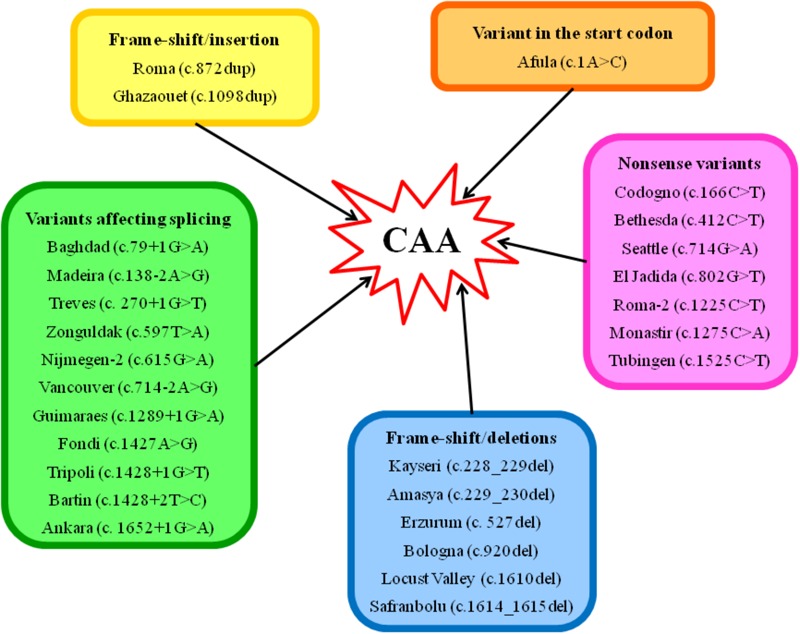
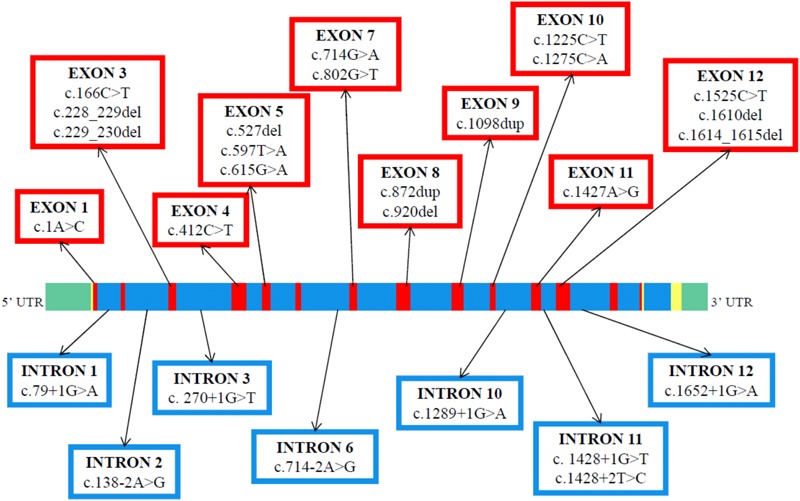
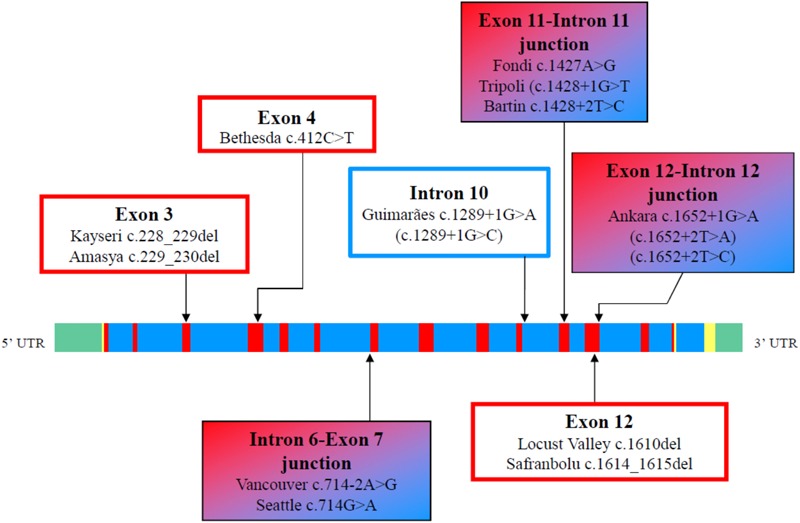
Congenital analbuminemia (CAA) is an inherited, autosomal recessive disorder with an incidence of 1:1,000,000 live birth. Affected individuals have a strongly decreased concentration, or complete absence, of serum albumin. The trait is usually detected by serum protein electrophoresis and immunochemistry techniques. However, due to the existence of other conditions in which the albumin concentrations are very low or null, analysis of the albumin (ALB) gene is necessary for the molecular diagnosis. CAA can lead to serious consequences in the prenatal period, because it can cause miscarriages and preterm birth, which often is due to oligohydramnios and placental abnormalities. Neonatally and in early childhood the trait is a risk factor that can lead to death, mainly from fluid retention and infections in the lower respiratory tract. By contrast, CAA is better tolerated in adulthood. Clinically, in addition to the low level of albumin, the patients almost always have hyperlipidemia, but they usually also have mild oedema, reduced blood pressure and fatigue. The fairly mild symptoms in adulthood are due to compensatory increment of other plasma proteins. The condition is rare; clinically, only about 90 cases have been detected worldwide. Among these, 53 have been studied by sequence analysis of the ALB gene, allowing the identification of 27 different loss of function (LoF) pathogenic variants. These include a variant in the start codon, frame-shift/insertions, frame-shift/deletions, nonsense variants, and variants affecting splicing. Most are unique, peculiar for each affected family, but one, a frame-shift deletion called Kayseri, has been found to cause about one third of the known cases allowing to presume a founder effect. This review provides an overview of the literature about CAA, about supportive and additional physiological and pharmacological information obtained from albumin-deficient mouse and rat models and a complete and up-to-date dataset of the pathogenic variants identified in the ALB gene.
Keywords: DNA-sequencing; analbuminemia; autosomal recessive; compensatory mechanisms; frequency; hyperlipidemia; pathogenic variations; preterm birth.
Figures
Similar articles
-
Severe hypercholesterolemia in a patient with very low albumin and normal renal function.J Clin Lipidol. 2023 Jan-Feb;17(1):64-67. doi: 10.1016/j.jacl.2022.10.011. Epub 2022 Nov 10. J Clin Lipidol. 2023. PMID: 36411186
-
A novel insertion (c.1098dupT) in the albumin gene causes analbuminemia in a consanguineous family.Eur J Med Genet. 2019 Feb;62(2):144-148. doi: 10.1016/j.ejmg.2018.07.001. Epub 2018 Jul 5. Eur J Med Genet. 2019. PMID: 29981851
-
A nucleotide deletion and frame-shift cause analbuminemia in a Turkish family.Biochem Med (Zagreb). 2016;26(2):264-71. doi: 10.11613/BM.2016.031. Biochem Med (Zagreb). 2016. PMID: 27346974 Free PMC article.
-
Congenital analbuminaemia: molecular defects and biochemical and clinical aspects.Biochim Biophys Acta. 2013 Dec;1830(12):5494-502. doi: 10.1016/j.bbagen.2013.04.019. Epub 2013 Apr 21. Biochim Biophys Acta. 2013. PMID: 23612153 Review.
-
The molecular genetic basis and diagnosis of familial hypercholesterolemia in Denmark.Dan Med Bull. 2002 Nov;49(4):318-45. Dan Med Bull. 2002. PMID: 12553167 Review.
Cited by
-
Role of sex hormone-binding globulin in the free hormone hypothesis and the relevance of free testosterone in androgen physiology.Cell Mol Life Sci. 2022 Oct 7;79(11):543. doi: 10.1007/s00018-022-04562-1. Cell Mol Life Sci. 2022. PMID: 36205798 Free PMC article. Review.
-
Incidental detection of hereditary bisalbuminemia in a patient with positive DAT coombs: A case-based review.Metabol Open. 2024 Jul 30;23:100307. doi: 10.1016/j.metop.2024.100307. eCollection 2024 Sep. Metabol Open. 2024. PMID: 39185032 Free PMC article. Review.
-
Is there reversible dimerization of albumin in blood plasma? And does it matter?Exp Physiol. 2024 Oct;109(10):1663-1671. doi: 10.1113/EP092012. Epub 2024 Aug 23. Exp Physiol. 2024. PMID: 39177455 Free PMC article. Review.
-
Albumin as a drug: its biological effects beyond volume expansion.Crit Care Resusc. 2020 Sep;22(3):257-265. doi: 10.1016/S1441-2772(23)00394-0. Crit Care Resusc. 2020. PMID: 32900333 Free PMC article. Review.
-
Recurrent Hypoglycemia in a Case of Congenital Analbuminemia.Case Rep Endocrinol. 2020 Feb 27;2020:8452564. doi: 10.1155/2020/8452564. eCollection 2020. Case Rep Endocrinol. 2020. PMID: 32181025 Free PMC article.
References
-
- Bennhold H. H., Peters H., Roth E. (1954). Uber einen fall von kompletter analbuminaemie ohne wesentliche klinische krankheitszichen. Verh. Dtsch. Ges. Inn. Med. 60 630–634. 10.1007/978-3-642-53819-3_139 - DOI
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous