

Genetic analysis of ATP7B in 102 south Indian families with Wilson disease
- PMID: 31059521
- PMCID: PMC6502322
- DOI: 10.1371/journal.pone.0215779
Genetic analysis of ATP7B in 102 south Indian families with Wilson disease
Abstract
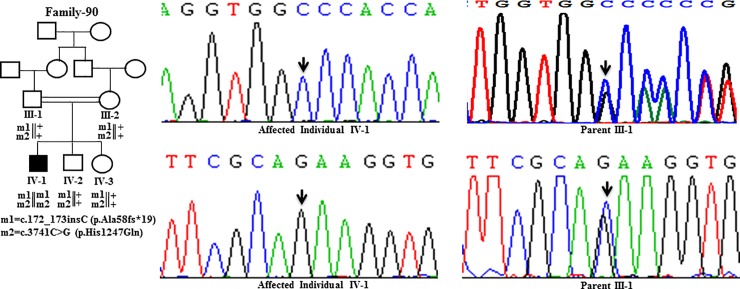
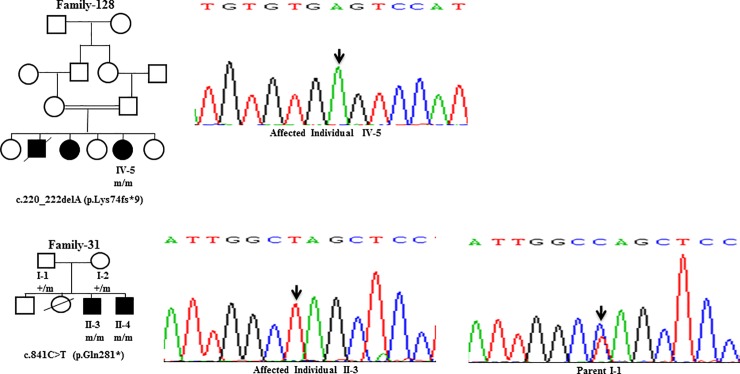
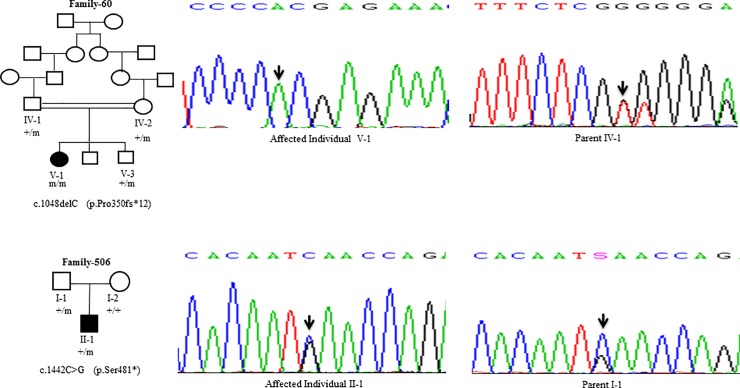
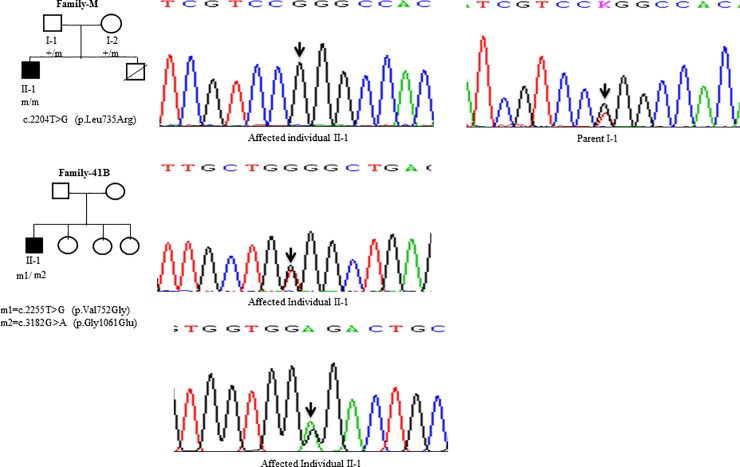
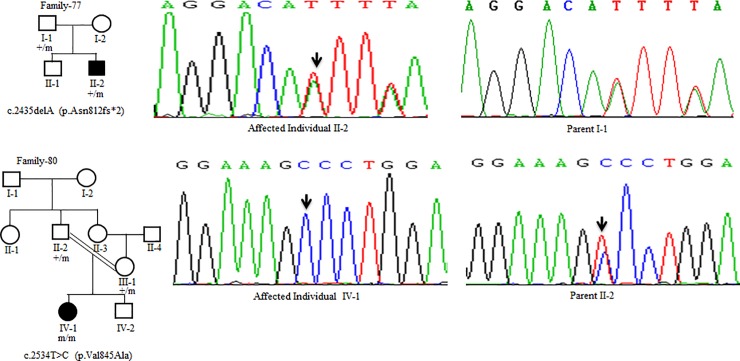
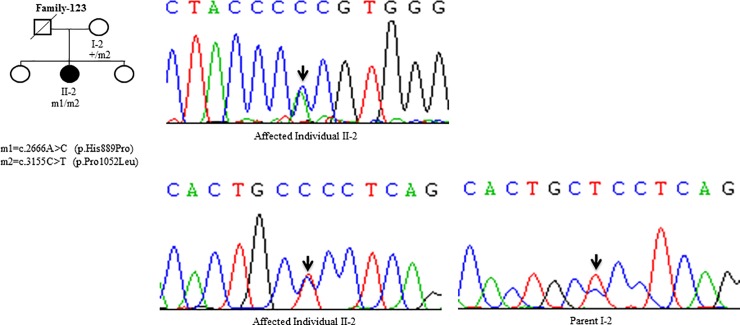
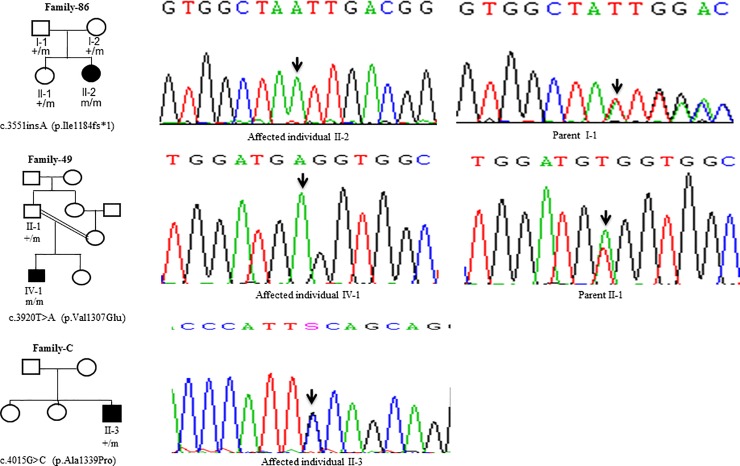
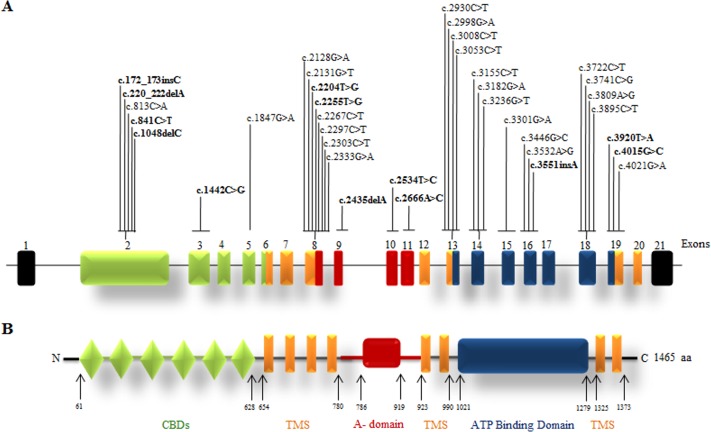
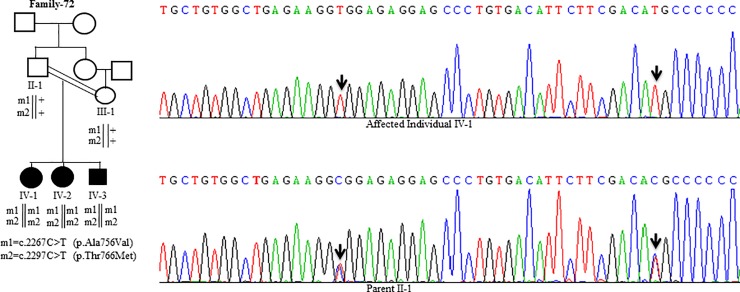
Wilson disease (WD) is an autosomal recessive disorder, characterized by excessive deposition of copper in various parts of the body, mainly in the liver and brain. It is caused by mutations in ATP7B. We report here the genetic analysis of 102 WD families from a south Indian population. Thirty-six different ATP7B mutations, including 13 novel ones [p.Ala58fs*19, p.Lys74fs*9, p.Gln281*, p.Pro350fs*12, p.Ser481*, p.Leu735Arg, p.Val752Gly, p.Asn812fs*2, p.Val845Ala, p.His889Pro, p.Ile1184fs*1, p.Val1307Glu and p.Ala1339Pro], were identified in 76/102 families. Interestingly, the mutation analysis of affected individuals in two families identified two different homozygous mutations in each family, and thus each affected individual from these families harbored two mutations in each ATP7B allele. Of 36 mutations, 28 were missense, thus making them the most prevalent mutations identified in the present study. Nonsense, insertion and deletion represented 3/36, 2/36 and 3/36 mutations, respectively. The haplotype analysis suggested founder effects for all the 14 recurrent mutations. Our study thus expands the mutational landscape of ATP7B with a total number of 758 mutations. The mutations identified during the present study will facilitate carrier and pre-symptomatic detection, and prenatal genetic diagnosis in affected families.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
