

Efficient integration of heterogeneous single-cell transcriptomes using Scanorama
- PMID: 31061482
- PMCID: PMC6551256
- DOI: 10.1038/s41587-019-0113-3
Efficient integration of heterogeneous single-cell transcriptomes using Scanorama
Abstract
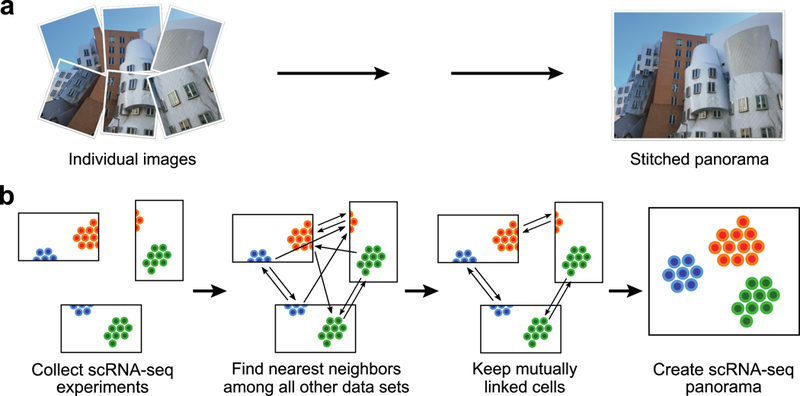
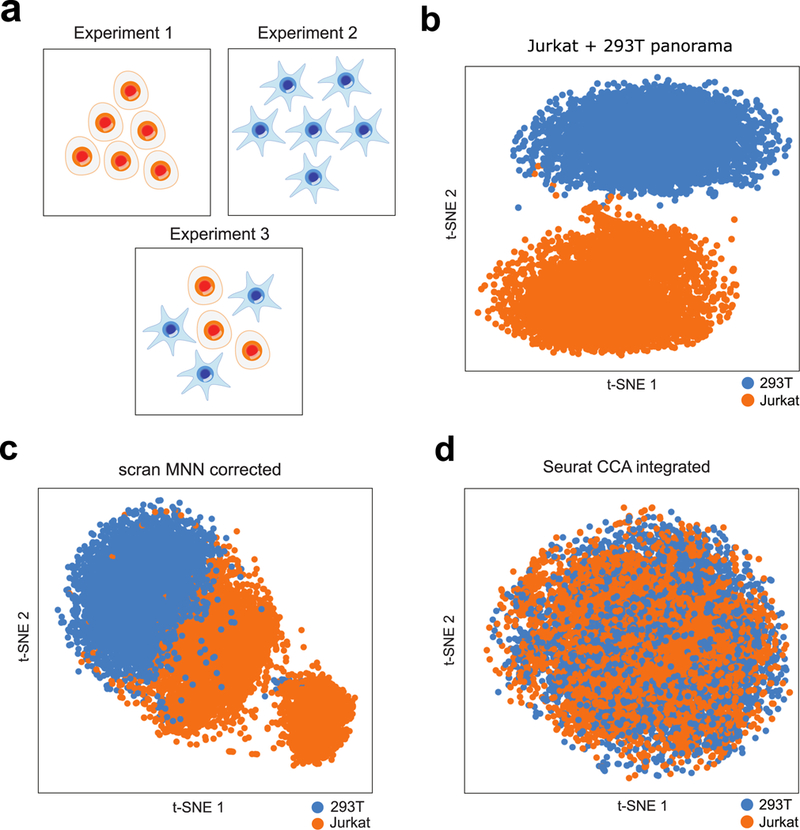
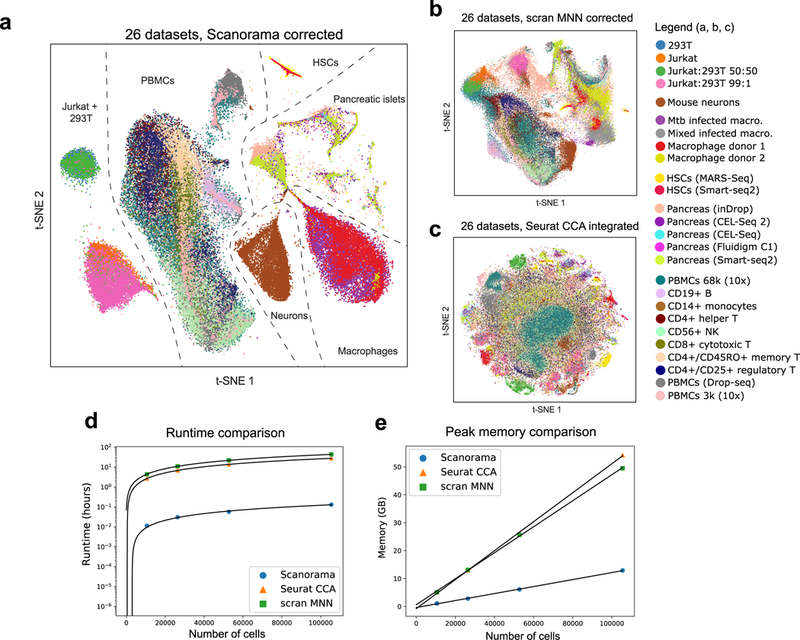
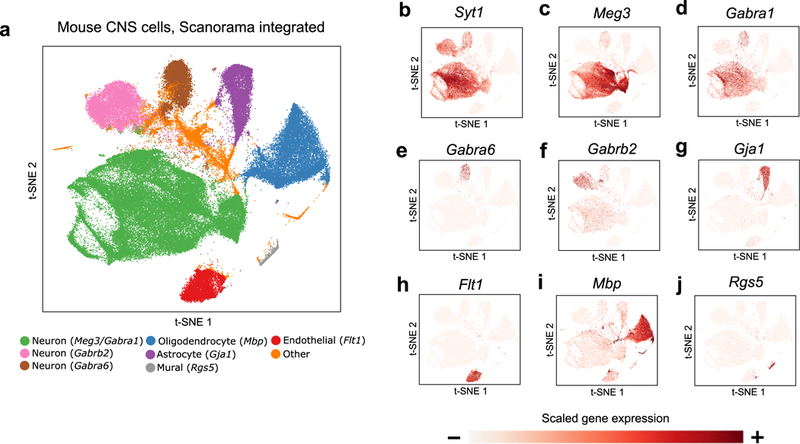
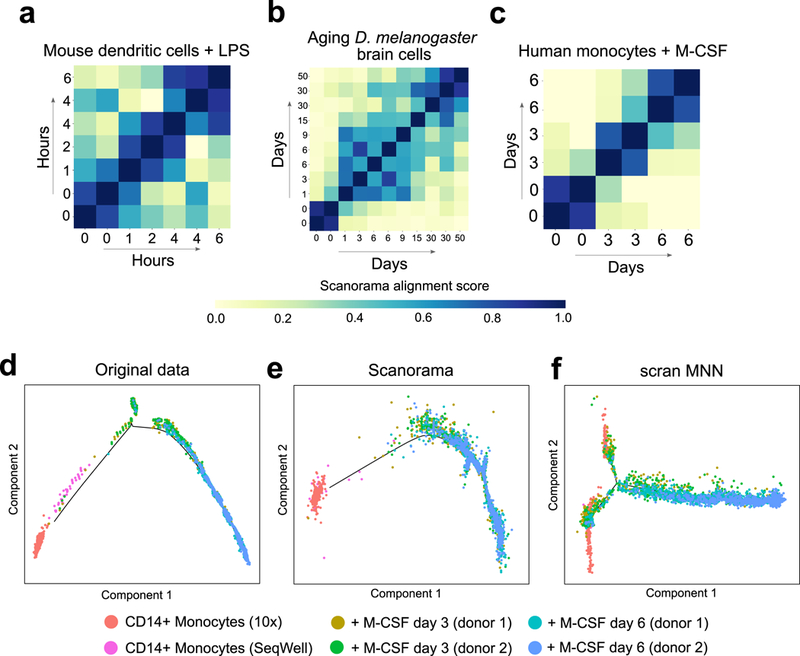
Integration of single-cell RNA sequencing (scRNA-seq) data from multiple experiments, laboratories and technologies can uncover biological insights, but current methods for scRNA-seq data integration are limited by a requirement for datasets to derive from functionally similar cells. We present Scanorama, an algorithm that identifies and merges the shared cell types among all pairs of datasets and accurately integrates heterogeneous collections of scRNA-seq data. We applied Scanorama to integrate and remove batch effects across 105,476 cells from 26 diverse scRNA-seq experiments representing 9 different technologies. Scanorama is sensitive to subtle temporal changes within the same cell lineage, successfully integrating functionally similar cells across time series data of CD14+ monocytes at different stages of differentiation into macrophages. Finally, we show that Scanorama is orders of magnitude faster than existing techniques and can integrate a collection of 1,095,538 cells in just ~9 h.
Figures
References
-
- Grün D et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 525, 251–255 (2015). - PubMed
Methods Only References
-
- Oliphant TE SciPy: Open source scientific tools for Python. Comput. Sci. Eng. 9, 10–20 (2007).
-
- Loh PR, Baym M & Berger B Compressive genomics. Nature Biotechnology 30, 627–630 (2012). - PubMed
-
- Van Der Maaten LJP & Hinton GE Visualizing high-dimensional data using t-SNE.J. Mach. Learn. Res. 9, 2579–2605 (2008).
-
- Pedregosa F & Varoquaux G Scikit-learn: Machine learning in Python. Journal of Machine Learning Research 12 (2011).
-
- Buttner M, Miao Z, Wolf A, Teichmann SA & Theis FJ A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 16, 43–49 (2017). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
