

Characterization of a Unique Form of Arrhythmic Cardiomyopathy Caused by Recessive Mutation in LEMD2
- PMID: 31061923
- PMCID: PMC6488817
- DOI: 10.1016/j.jacbts.2018.12.001
Characterization of a Unique Form of Arrhythmic Cardiomyopathy Caused by Recessive Mutation in LEMD2
Abstract
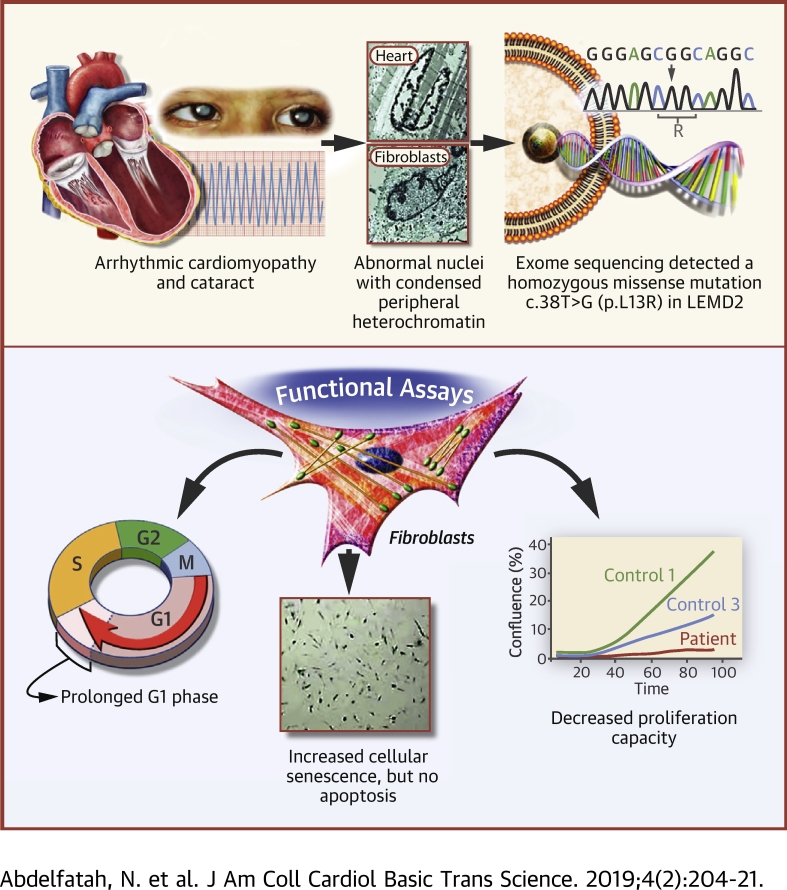
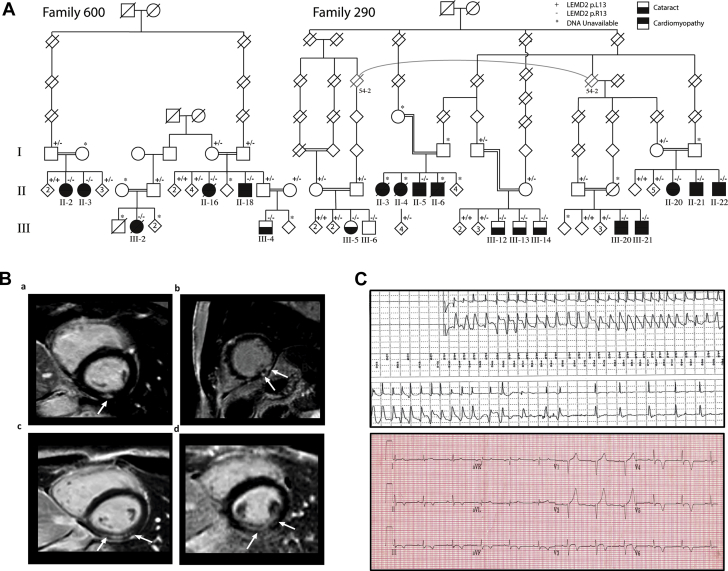
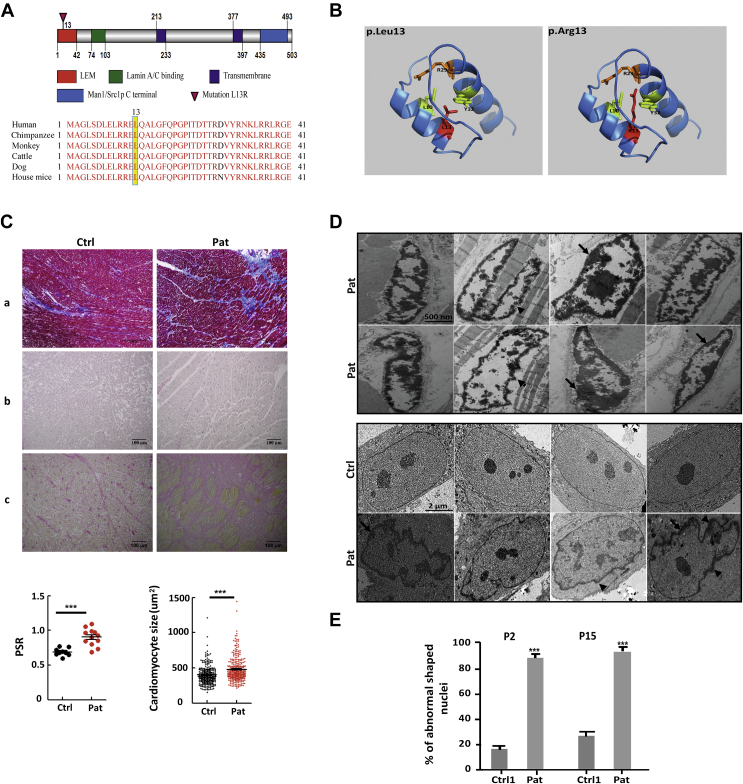
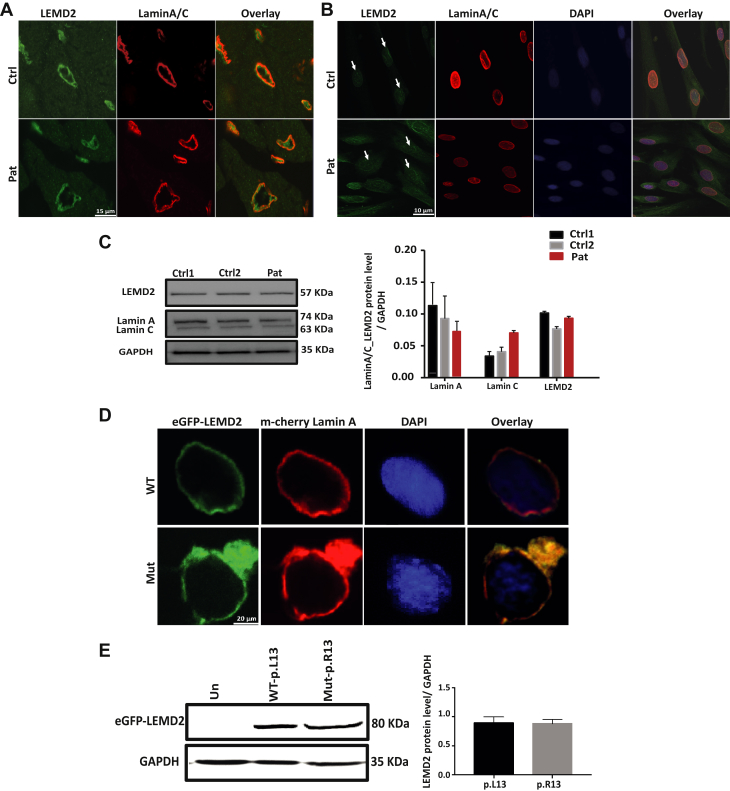
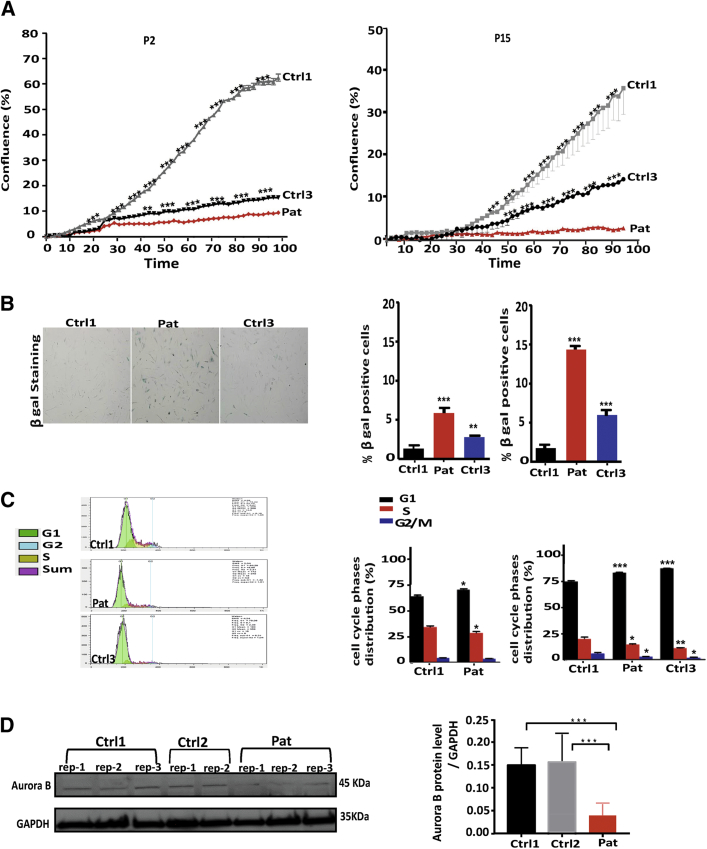
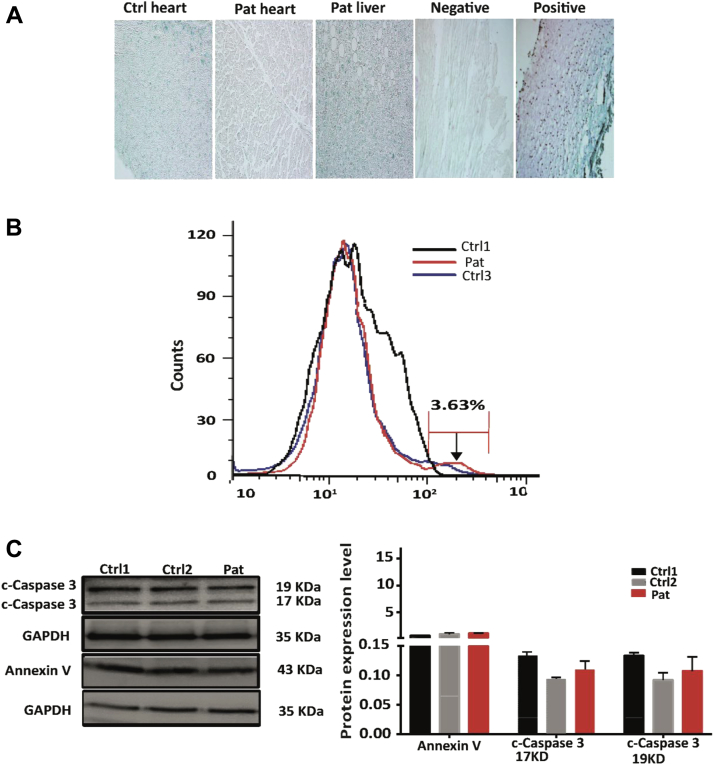
Nuclear envelope proteins have been shown to play an important role in the pathogenesis of inherited dilated cardiomyopathy. Here, we present a remarkable cardiac phenotype caused by a homozygous LEMD2 mutation in patients of the Hutterite population with juvenile cataract. Mutation carriers develop arrhythmic cardiomyopathy with mild impairment of left ventricular systolic function but severe ventricular arrhythmias leading to sudden cardiac death. Affected cardiac tissue from a deceased patient and fibroblasts exhibit elongated nuclei with abnormal condensed heterochromatin at the periphery. The patient fibroblasts demonstrate cellular senescence and reduced proliferation capacity, which may suggest an involvement of LEM domain containing protein 2 in chromatin remodeling processes and premature aging.
Keywords: ACM, arrhythmogenic cardiomyopathy; BANF, barrier to autointegration factor; CMR, cardiac magnetic resonance; DAPI, 4′,6′-diamidino-2-phenylindole; DCM, dilated cardiomyopathy; DNA, deoxyribonucleic acid; EMD, emerin; ICD, implantable cardioverter-defibrillator; LEMD2; LEMD2, LEM domain containing protein 2; LGE, late gadolinium enhancement; LMNA, lamin A/C; LV, left ventricular; NE, nuclear envelope; P, passage; PBS, phosphate-buffered saline; SAHF, senescence-associated heterochromatin foci; SNV, single nucleotide variant; chromatin remodeling; dilated cardiomyopathy; eGFP, enhanced green fluorescent protein; inner nuclear membrane; sudden death.
Figures
References
-
- Haas J., Frese K.S., Peil B. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36:1123–1135. - PubMed
-
- Fatkin D., MacRae C., Sasaki T. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. - PubMed
-
- Bione S., Maestrini E., Rivella S. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–327. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
