

Improved annotation of the domestic pig genome through integration of Iso-Seq and RNA-seq data
- PMID: 31064321
- PMCID: PMC6505119
- DOI: 10.1186/s12864-019-5709-y
Improved annotation of the domestic pig genome through integration of Iso-Seq and RNA-seq data
Abstract
Background: Our understanding of the pig transcriptome is limited. RNA transcript diversity among nine tissues was assessed using poly(A) selected single-molecule long-read isoform sequencing (Iso-seq) and Illumina RNA sequencing (RNA-seq) from a single White cross-bred pig.
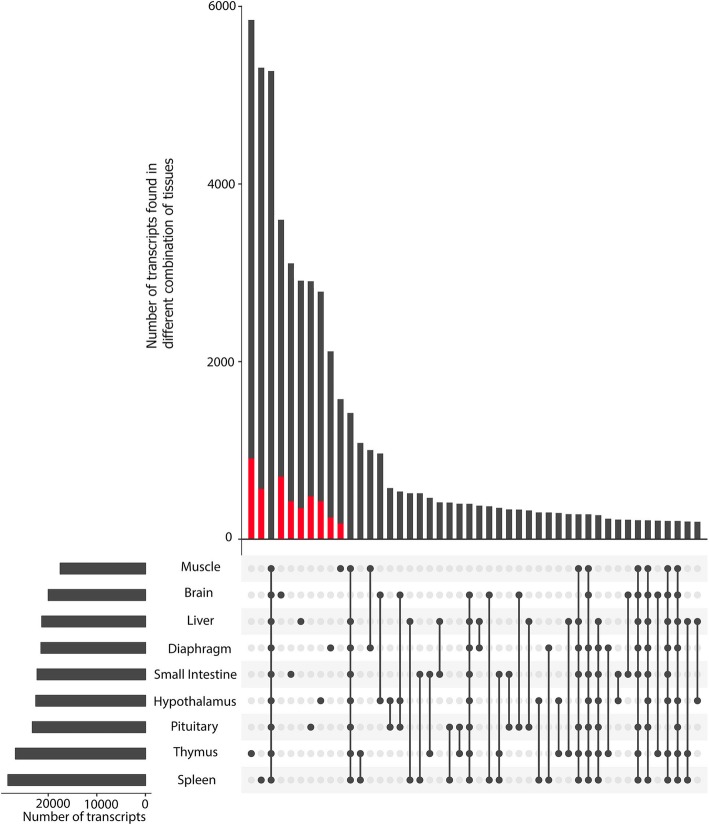
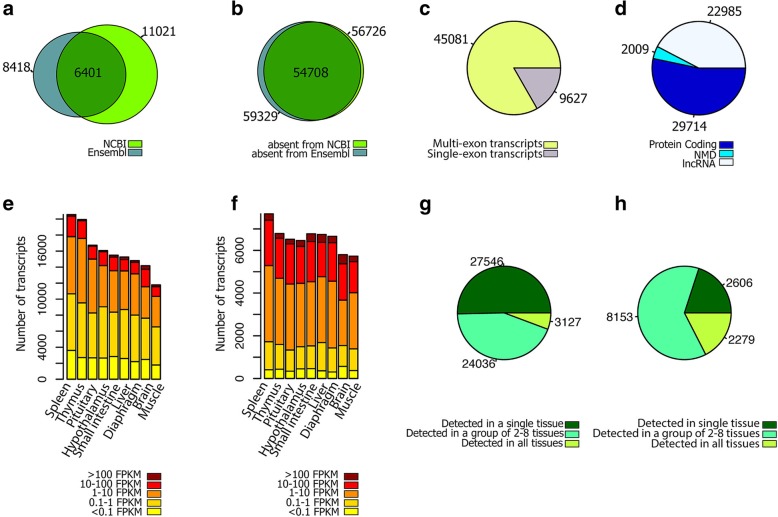
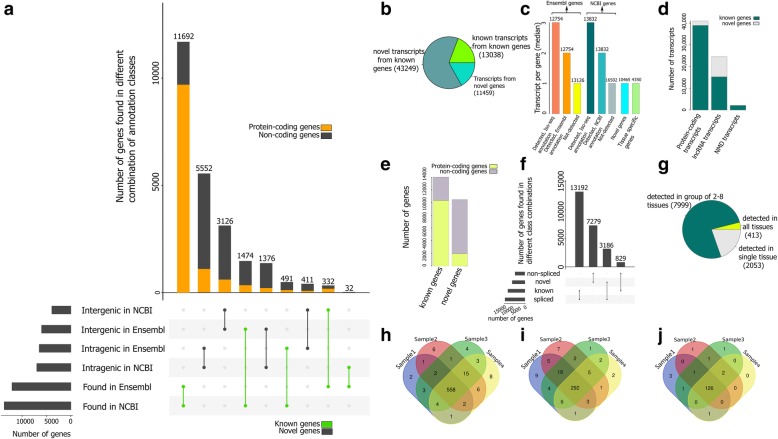
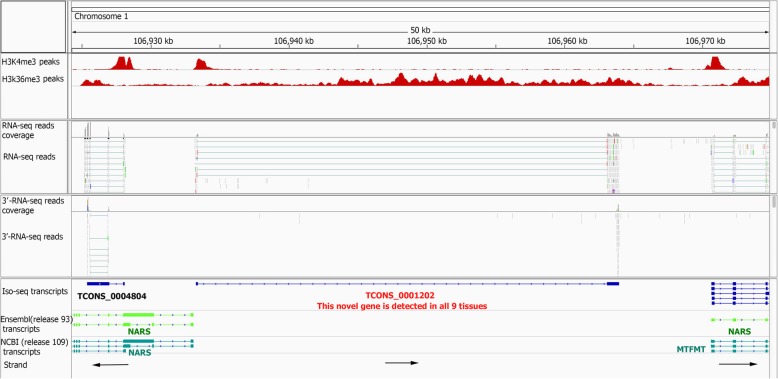
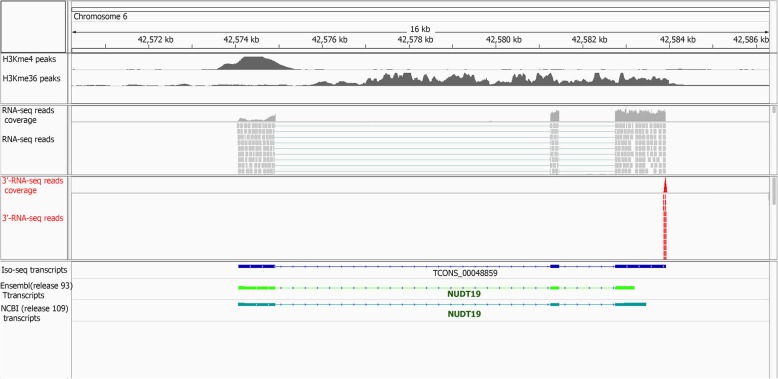
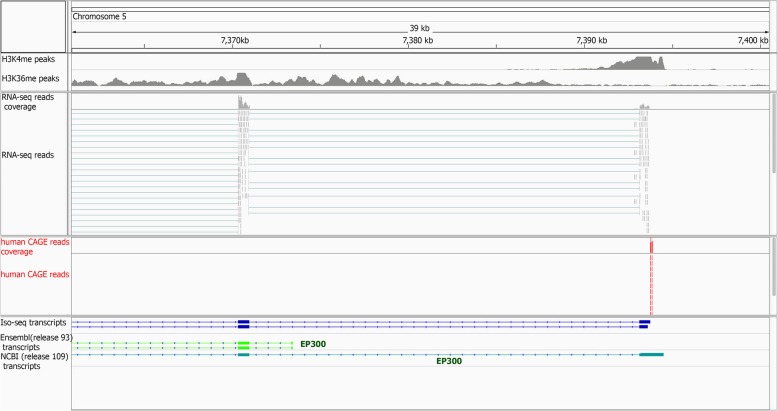
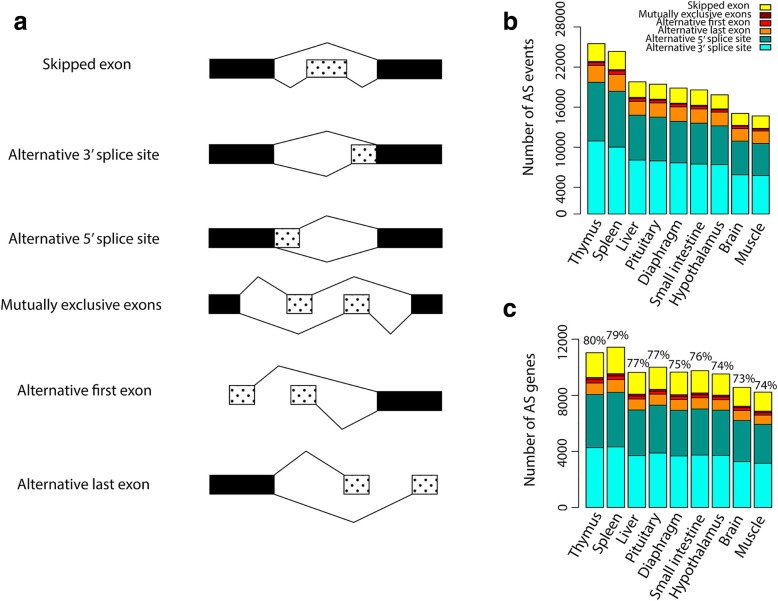
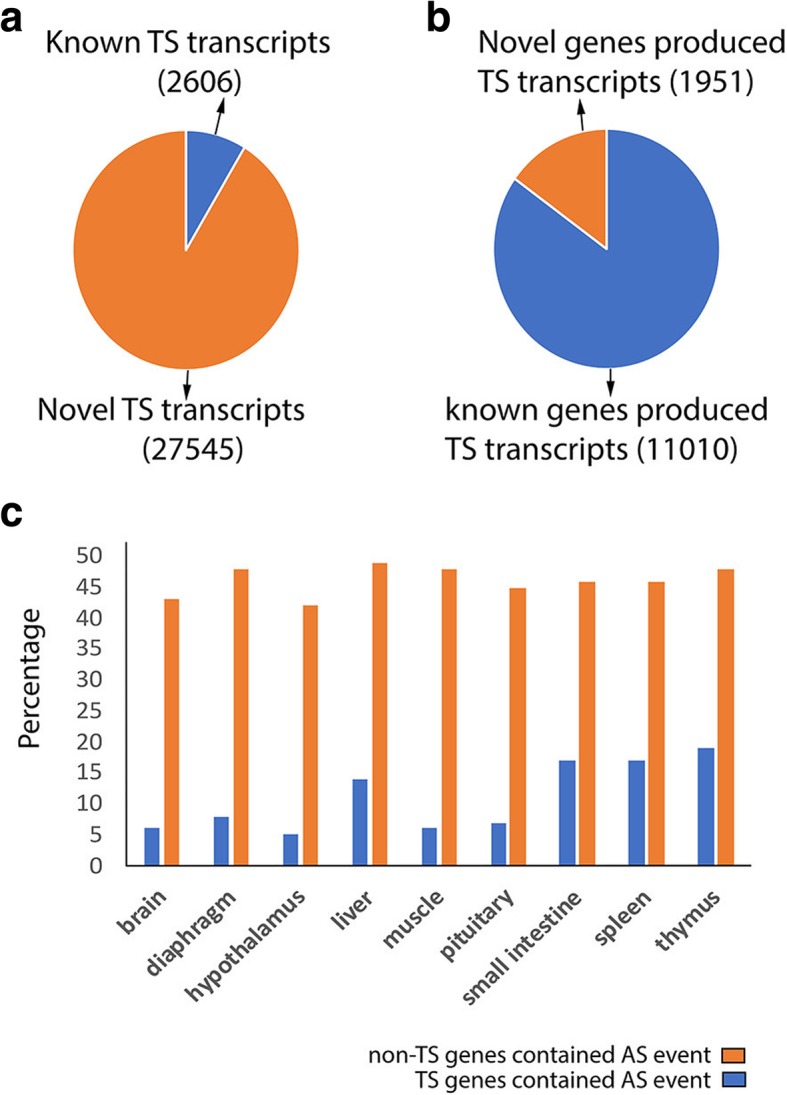
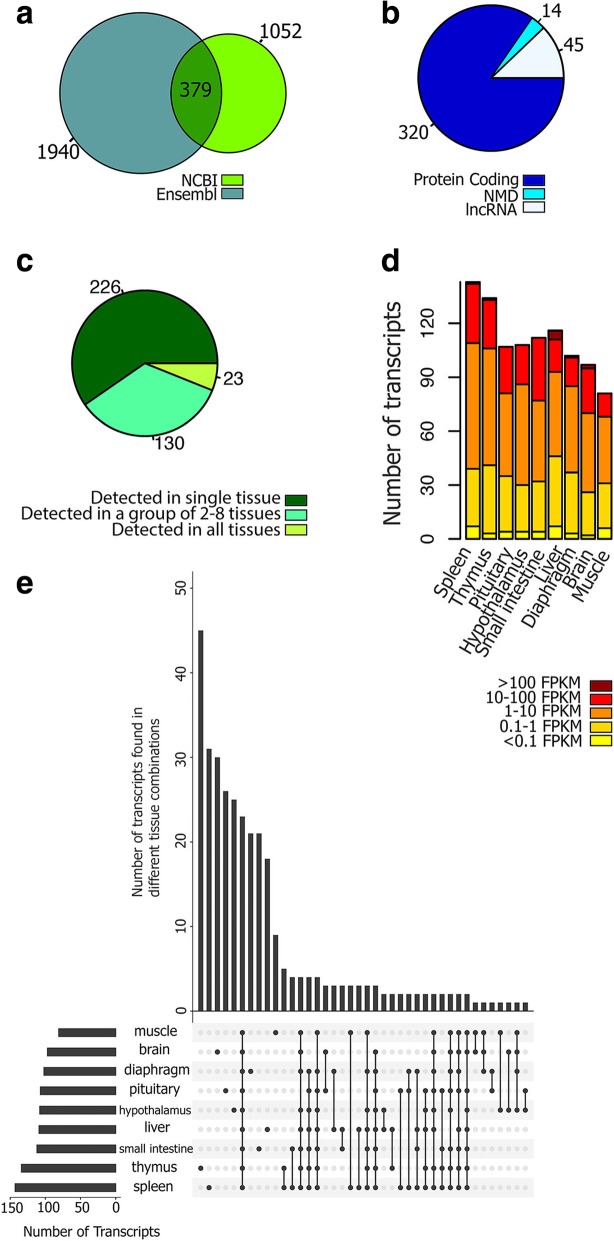
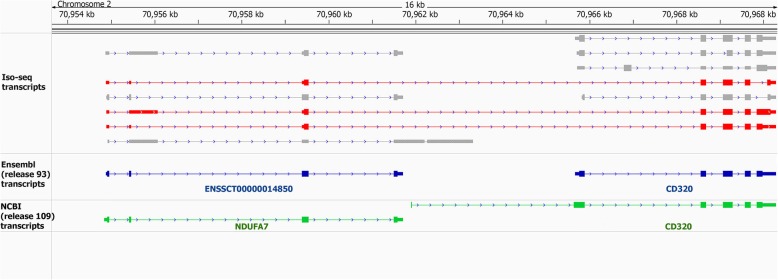
Results: Across tissues, a total of 67,746 unique transcripts were observed, including 60.5% predicted protein-coding, 36.2% long non-coding RNA and 3.3% nonsense-mediated decay transcripts. On average, 90% of the splice junctions were supported by RNA-seq within tissue. A large proportion (80%) represented novel transcripts, mostly produced by known protein-coding genes (70%), while 17% corresponded to novel genes. On average, four transcripts per known gene (tpg) were identified; an increase over current EBI (1.9 tpg) and NCBI (2.9 tpg) annotations and closer to the number reported in human genome (4.2 tpg). Our new pig genome annotation extended more than 6000 known gene borders (5' end extension, 3' end extension, or both) compared to EBI or NCBI annotations. We validated a large proportion of these extensions by independent pig poly(A) selected 3'-RNA-seq data, or human FANTOM5 Cap Analysis of Gene Expression data. Further, we detected 10,465 novel genes (81% non-coding) not reported in current pig genome annotations. More than 80% of these novel genes had transcripts detected in > 1 tissue. In addition, more than 80% of novel intergenic genes with at least one transcript detected in liver tissue had H3K4me3 or H3K36me3 peaks mapping to their promoter and gene body, respectively, in independent liver chromatin immunoprecipitation data.
Conclusions: These validated results show significant improvement over current pig genome annotations.
Keywords: Genome annotation; Iso-seq; PacBio; Porcine; RNA-seq; Single molecule long read sequencing; Transcriptome sequencing.
Conflict of interest statement
Ethics approval and consent to participate
The cross-bred pig used for genome sequencing, and transcriptome sequencing by both PacBio IsoSeq and Illumina RNA-seq technologies were from USMARC. Protocols for use, care and handling pigs were approved by IACUCs at Iowa State University or USMARC. Pigs used to generate the unpublished sequencing data were maintained in Iowa State University or USMARC.
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Thibaud-Nissen F SA, Murphy T, et al. The Eukaryotic Genome Annotation Pipeline. 2013 Nov 14. In: The NCBI Handbook [Internet]. 2nd edition. Bethesda (MD): National Center for Biotechnology Information (US); 2013-. Available from: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/. Accessed 14 Nov 2013.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
