

Disruption of asxl1 results in myeloproliferative neoplasms in zebrafish
- PMID: 31064769
- PMCID: PMC6550042
- DOI: 10.1242/dmm.035790
Disruption of asxl1 results in myeloproliferative neoplasms in zebrafish
Abstract
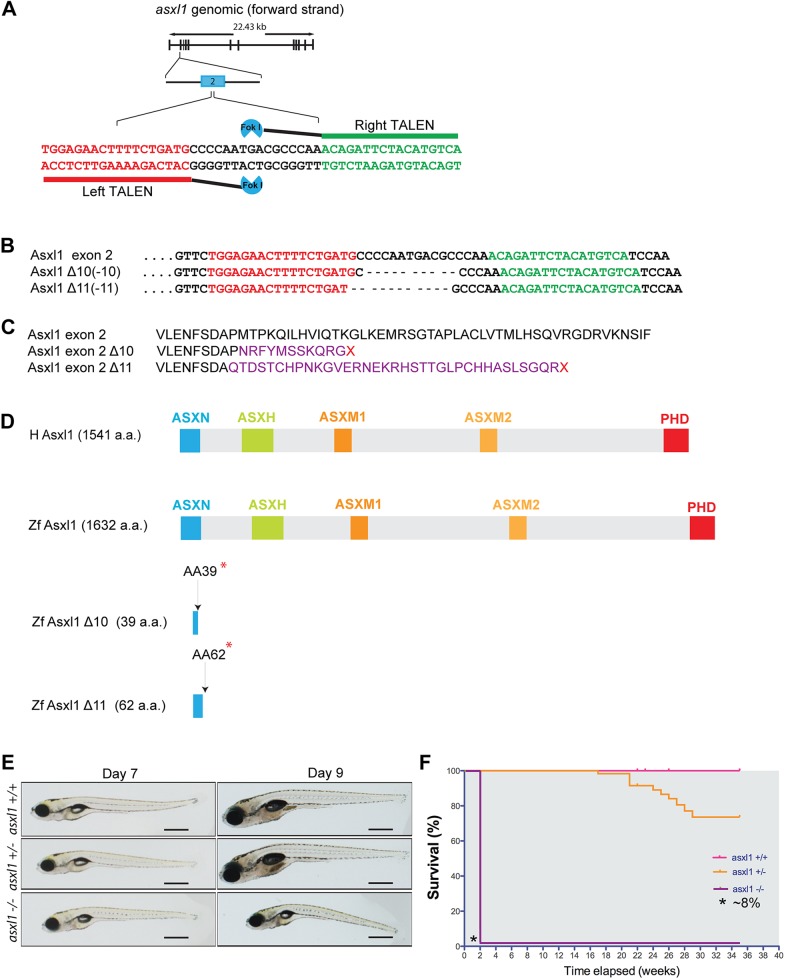
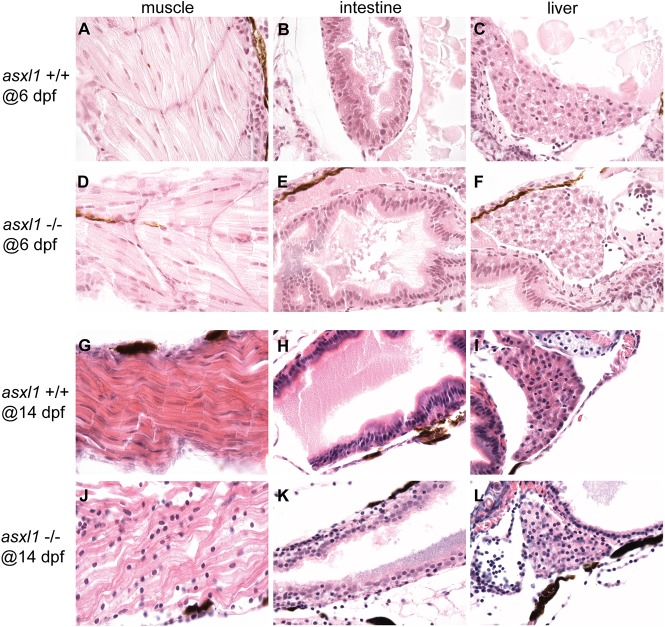
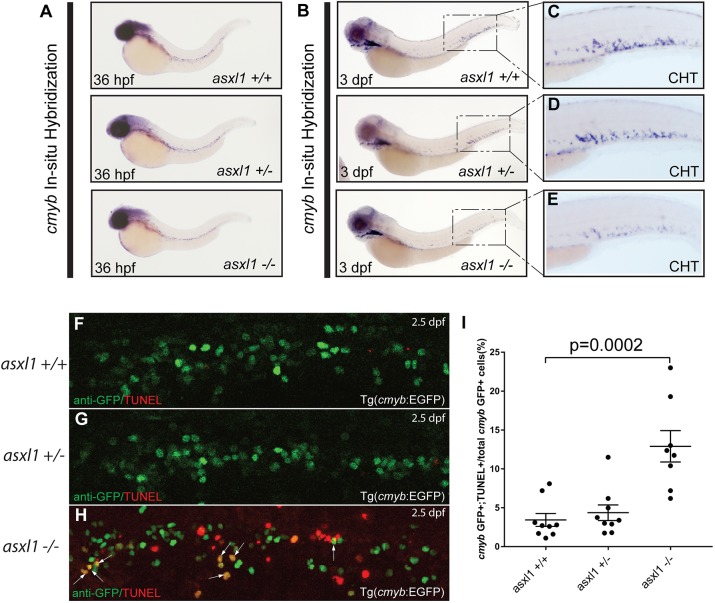
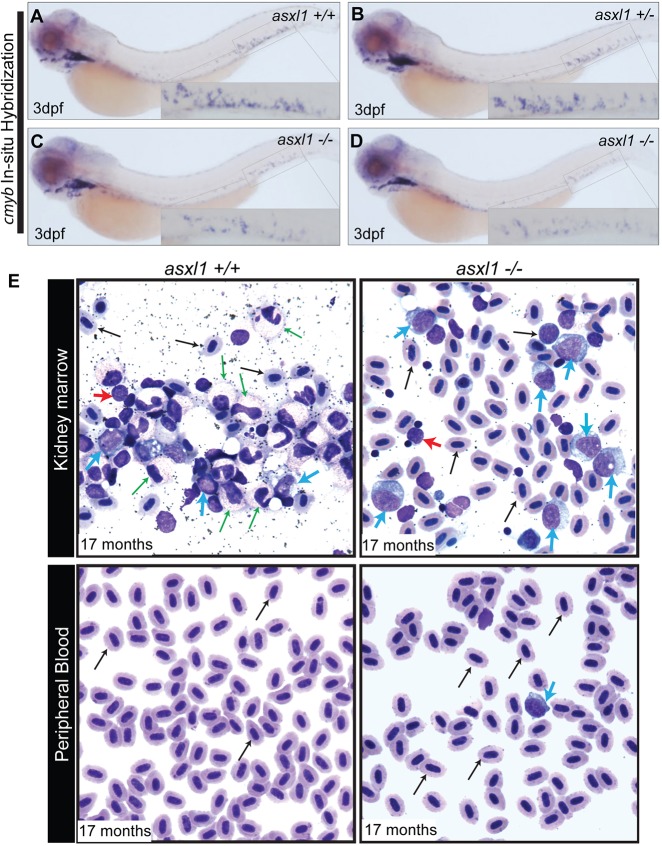
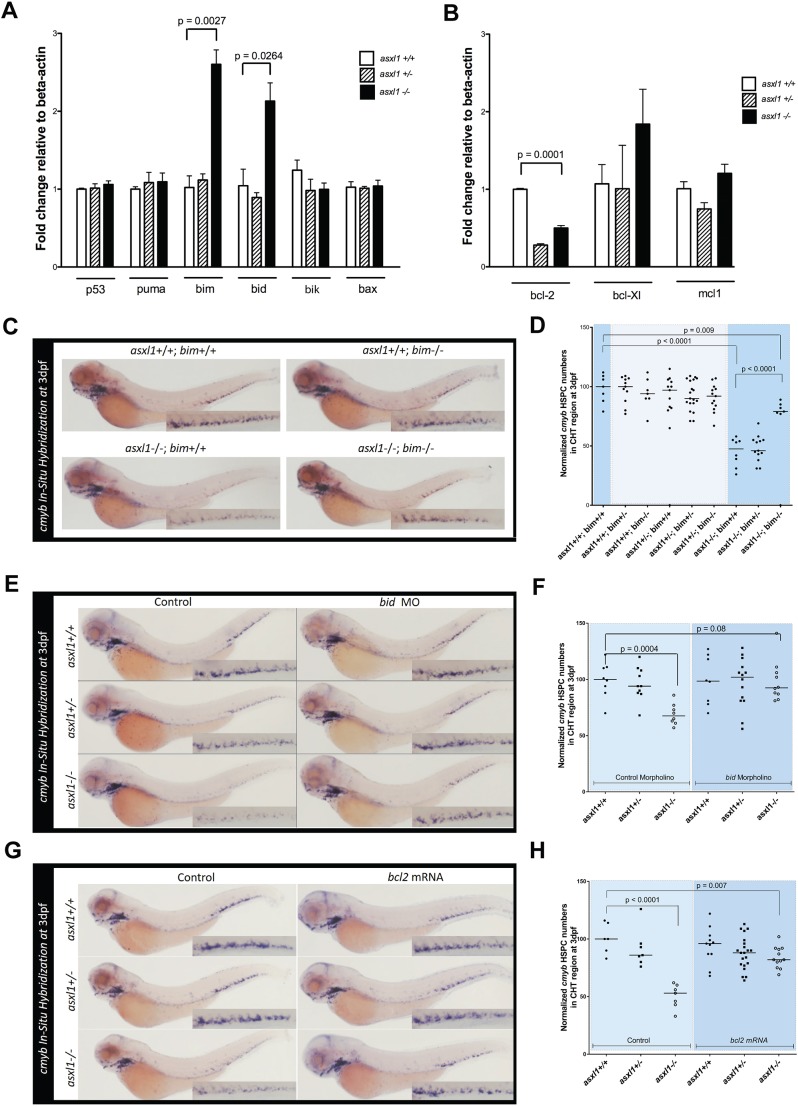
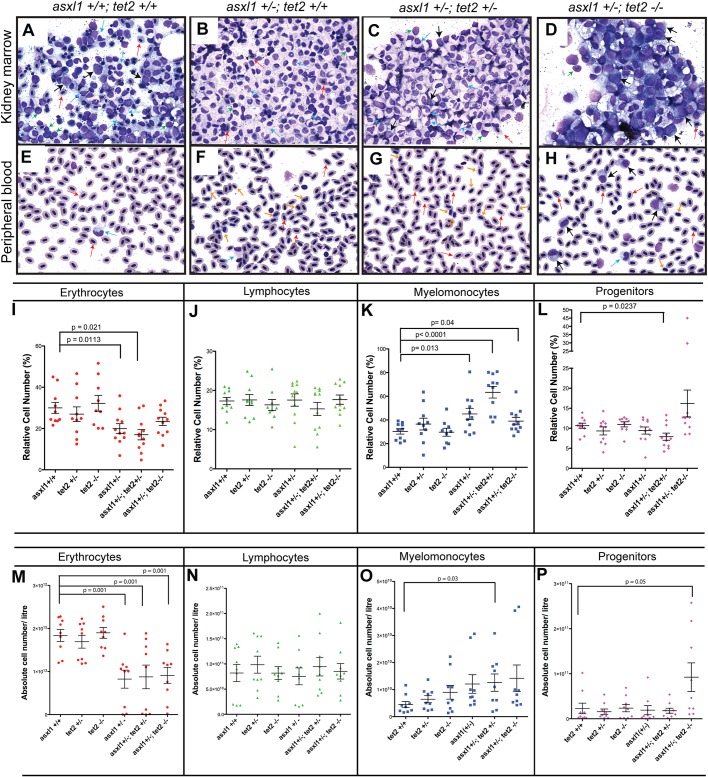
Somatic loss-of-function mutations of the additional sex combs-like transcriptional regulator 1 (ASXL1) gene are common genetic abnormalities in human myeloid malignancies and induce clonal expansion of mutated hematopoietic stem cells (HSCs). To understand how ASXL1 disruption leads to myeloid cell transformation, we generated asxl1 haploinsufficient and null zebrafish lines using genome-editing technology. Here, we show that homozygous loss of asxl1 leads to apoptosis of newly formed HSCs. Apoptosis occurred via the mitochondrial apoptotic pathway mediated by upregulation of bim and bid Half of the asxl1+/- zebrafish had myeloproliferative neoplasms (MPNs) by 5 months of age. Heterozygous loss of asxl1 combined with heterozygous loss of tet2 led to a more penetrant MPN phenotype, while heterozygous loss of asxl1 combined with complete loss of tet2 led to acute myeloid leukemia (AML). These findings support the use of asxl1+/- zebrafish as a strategy to identify small-molecule drugs to suppress the growth of asxl1 mutant but not wild-type HSCs in individuals with somatically acquired inactivating mutations of ASXL1.
Keywords: Apoptosis; Genome editing; Hematopoietic stem cells; Myeloproliferative neoplasms; Tet2.
© 2019. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interestsK.J. has financial interests in Beam Therapeutics, Editas Medicine, Endcadia, Pairwise Plants, Poseida Therapeutics, and Transposagen Biopharmaceuticals; holds equity in Excelsior Genomics; is a member of the Board of Directors of the American Society of Gene and Cell Therapy; and is a co-inventor on various patents and patent applications that describe gene editing and epigenetic editing technologies. K.J.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. The other authors have no competing or financial interests to declare.
Figures
References
-
- Abdel-Wahab O., Pardanani A., Patel J., Wadleigh M., Lasho T., Heguy A., Beran M., Gilliland D. G., Levine R. L. and Tefferi A. (2011). Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia 25, 1200-1202. 10.1038/leu.2011.58 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
