

SWINGER: a clustering algorithm for concurrent coupling of atomistic and supramolecular liquids
- PMID: 31065343
- PMCID: PMC6501350
- DOI: 10.1098/rsfs.2018.0075
SWINGER: a clustering algorithm for concurrent coupling of atomistic and supramolecular liquids
Abstract
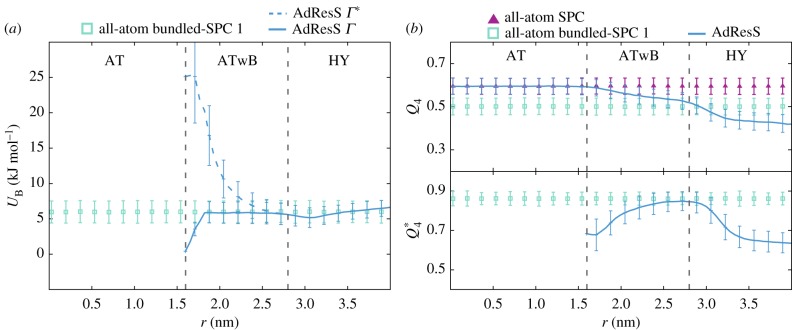
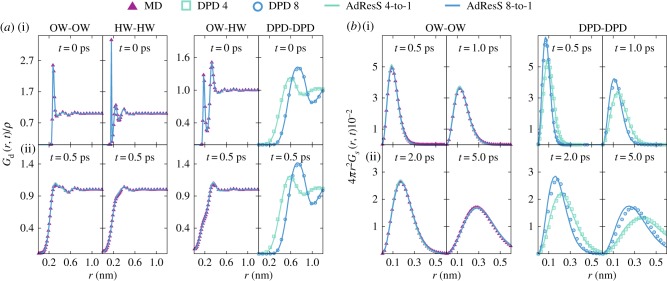
In this contribution, we review recent developments and applications of a dynamic clustering algorithm SWINGER tailored for the multiscale molecular simulations of biomolecular systems. The algorithm on-the-fly redistributes solvent molecules among supramolecular clusters. In particular, we focus on its applications in combination with the adaptive resolution scheme, which concurrently couples atomistic and coarse-grained molecular representations. We showcase the versatility of our multiscale approach on a few applications to biomolecular systems coupling atomistic and supramolecular water models such as the well-established MARTINI and dissipative particle dynamics models and provide an outlook for future work.
Keywords: adaptive resolution; molecular dynamics; supramolecular coupling.
Conflict of interest statement
We declare we have no competing interests.
Figures




References
-
- Allen MP, Tildesley DJ. 1989. Computer simulation of liquids. Oxford, UK: Clarendon Press.
-
- Tuckerman ME. 2010. Statistical mechanics: theory and molecular simulation. New York, NY: Oxford University Press.
Publication types
LinkOut - more resources
Full Text Sources
