

Illumina and Nanopore methods for whole genome sequencing of hepatitis B virus (HBV)
- PMID: 31068626
- PMCID: PMC6506499
- DOI: 10.1038/s41598-019-43524-9
Illumina and Nanopore methods for whole genome sequencing of hepatitis B virus (HBV)
Abstract
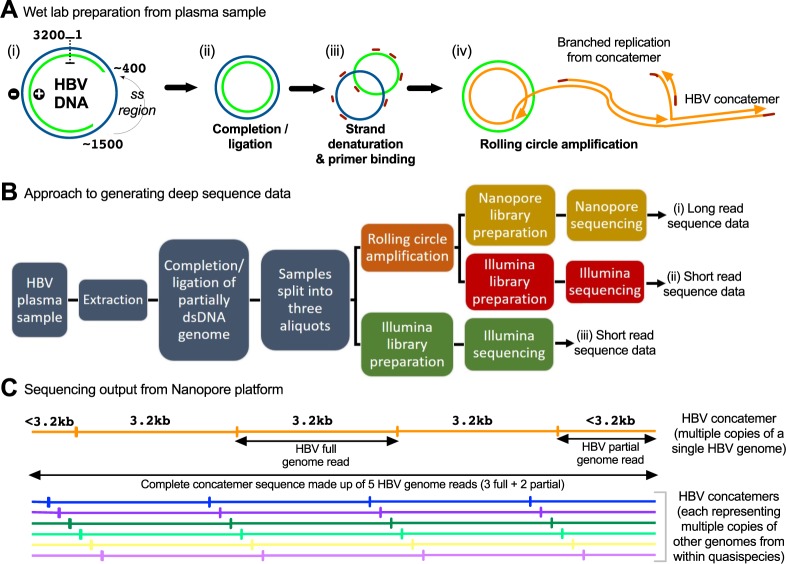
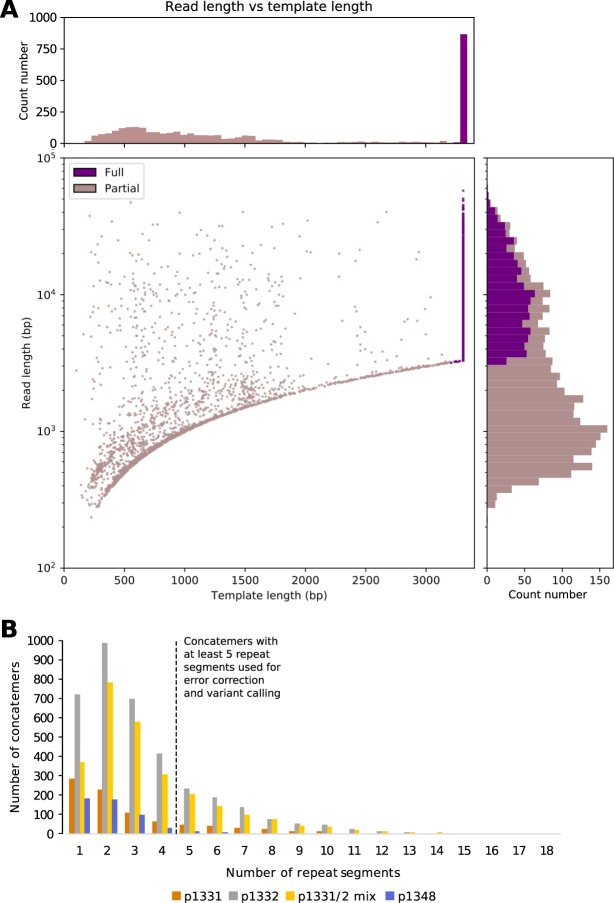
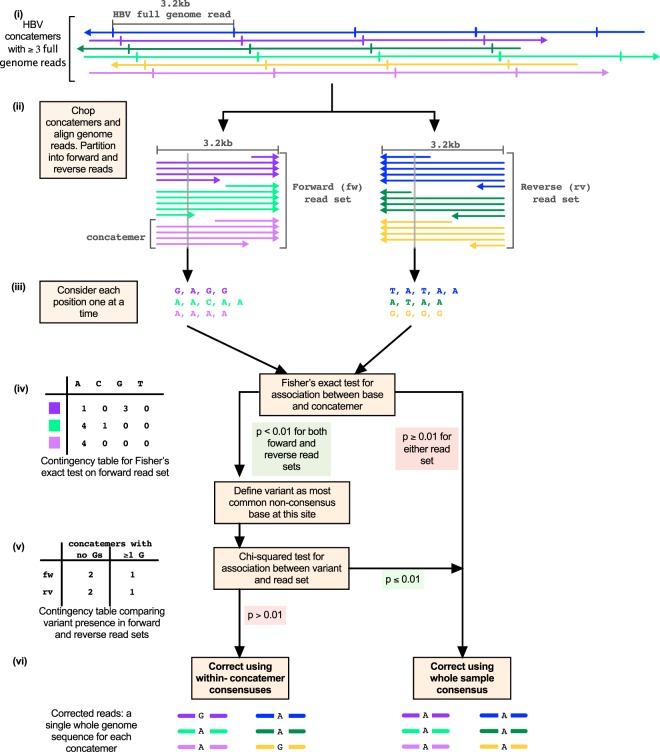
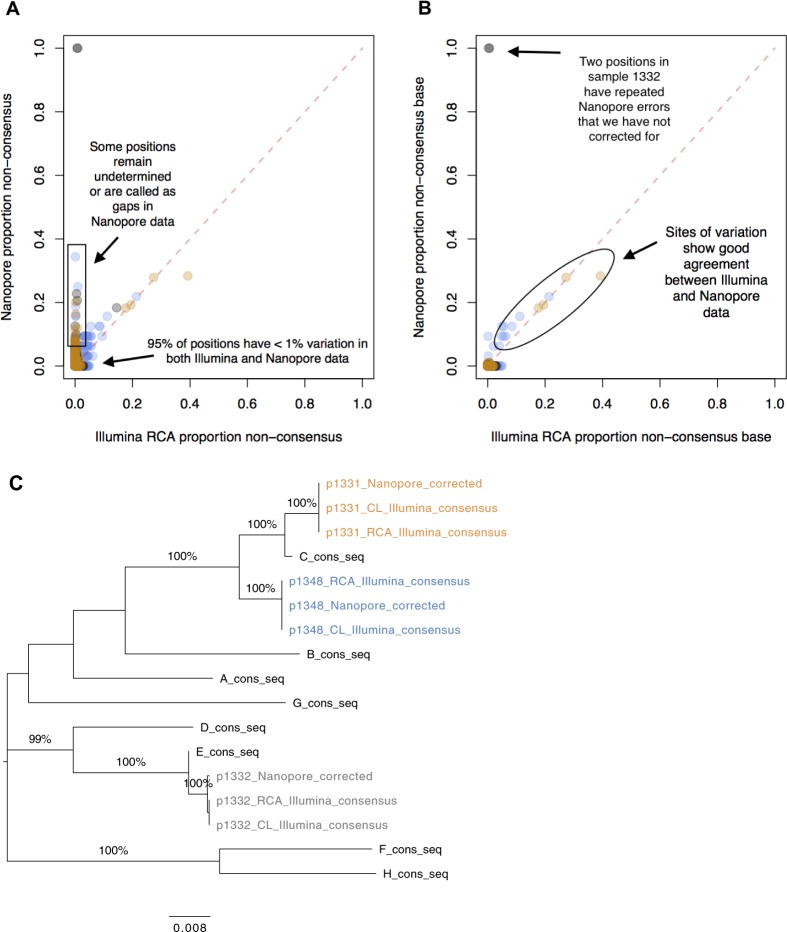
Advancing interventions to tackle the huge global burden of hepatitis B virus (HBV) infection depends on improved insights into virus epidemiology, transmission, within-host diversity, drug resistance and pathogenesis, all of which can be advanced through the large-scale generation of full-length virus genome data. Here we describe advances to a protocol that exploits the circular HBV genome structure, using isothermal rolling-circle amplification to enrich HBV DNA, generating concatemeric amplicons containing multiple successive copies of the same genome. We show that this product is suitable for Nanopore sequencing as single reads, as well as for generating short-read Illumina sequences. Nanopore reads can be used to implement a straightforward method for error correction that reduces the per-read error rate, by comparing multiple genome copies combined into a single concatemer and by analysing reads generated from plus and minus strands. With this approach, we can achieve an improved consensus sequencing accuracy of 99.7% and resolve intra-sample sequence variants to form whole-genome haplotypes. Thus while Illumina sequencing may still be the most accurate way to capture within-sample diversity, Nanopore data can contribute to an understanding of linkage between polymorphisms within individual virions. The combination of isothermal amplification and Nanopore sequencing also offers appealing potential to develop point-of-care tests for HBV, and for other viruses.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Razavi-Shearer Devin, Gamkrelidze Ivane, Nguyen Mindie H, Chen Ding-Shinn, Van Damme Pierre, Abbas Zaigham, Abdulla Maheeba, Abou Rached Antoine, Adda Danjuma, Aho Inka, Akarca Ulus, Hasan Fuad, Al Lawati Faryal, Al Naamani Khalid, Al-Ashgar Hamad Ibrahim, Alavian Seyed M, Alawadhi Sameer, Albillos Agustin, Al-Busafi Said A, Aleman Soo, Alfaleh Faleh Z, Aljumah Abdulrahman A, Anand Anil C, Anh Nguyen Thu, Arends Joop E, Arkkila Perttu, Athanasakis Kostas, Bane Abate, Ben-Ari Ziv, Berg Thomas, Bizri Abdul R, Blach Sarah, Brandão Mello Carlos E, Brandon Samantha M, Bright Bisi, Bruggmann Philip, Brunetto Maurizia, Buti Maria, Chan Henry L Y, Chaudhry Asad, Chien Rong-Nan, Choi Moon S, Christensen Peer B, Chuang Wan-Long, Chulanov Vladimir, Clausen Mette R, Colombo Massimo, Cornberg Markus, Cowie Benjamin, Craxi Antonio, Croes Esther A, Cuellar Diego Alberto, Cunningham Chris, Desalegn Hailemichael, Drazilova Sylvia, Duberg Ann-Sofi, Egeonu Steve S, El-Sayed Manal H, Estes Chris, Falconer Karolin, Ferraz Maria L G, Ferreira Paulo R, Flisiak Robert, Frankova Sona, Gaeta Giovanni B, García-Samaniego Javier, Genov Jordan, Gerstoft Jan, Goldis Adrian, Gountas Ilias, Gray Richard, Guimarães Pessôa Mário, Hajarizadeh Behzad, Hatzakis Angelos, Hézode Christophe, Himatt Sayed M, Hoepelman Andy, Hrstic Irena, Hui Yee-Tak T, Husa Petr, Jahis Rohani, Janjua Naveed Z, Jarčuška Peter, Jaroszewicz Jerzy, Kaymakoglu Sabahattin, Kershenobich David, Kondili Loreta A, Konysbekova Aliya, Krajden Mel, Kristian Pavol, Laleman Wim, Lao Wai-cheung C, Layden Jen, Lazarus Jeffrey V, Lee Mei-Hsuan, Liakina Valentina, Lim Young-Suk S, Loo Ching-kong K, Lukšić Boris, Malekzadeh Reza, Malu Abraham O, Mamatkulov Adkhamjon, Manns Michael, Marinho Rui T, Maticic Mojca, Mauss Stefan, Memon Muhammad S, Mendes Correa Maria C, Mendez-Sanchez Nahum, Merat Shahin, Metwally Ammal M, Mohamed Rosmawati, Mokhbat Jacques E, Moreno Christophe, Mossong Joel, Mourad Fadi H, Müllhaupt Beat, Murphy Kimberly, Musabaev Erkin, Nawaz Arif, Nde Helen M, Negro Francesco, Nersesov Alexander, Nguyen Van Thi Thuy, Njouom Richard, Ntagirabiri Renovat, Nurmatov Zuridin, Obekpa Solomon, Ocama Ponsiano, Oguche Stephen, Omede Ogu, Omuemu Casimir, Opare-Sem Ohene, Opio Christopher K, Örmeci Necati, Papatheodoridis George, Pasini Ken, Pimenov Nikolay, Poustchi Hossein, Quang Trân D, Qureshi Huma, Ramji Alnoor, Razavi-Shearer Kathryn, Redae Berhane, Reesink Henk W, Rios Cielo Yaneth, Rjaskova Gabriela, Robbins Sarah, Roberts Lewis R, Roberts Stuart K, Ryder Stephen D, Safadi Rifaat, Sagalova Olga, Salupere Riina, Sanai Faisal M, Sanchez-Avila Juan F, Saraswat Vivek, Sarrazin Christoph, Schmelzer Jonathan D, Schréter Ivan, Scott Julia, Seguin-Devaux Carole, Shah Samir R, Sharara Ala I, Sharma Manik, Shiha Gamal E, Shin Tesia, Sievert William, Sperl Jan, Stärkel Peter, Stedman Catherine, Sypsa Vana, Tacke Frank, Tan Soek S, Tanaka Junko, Tomasiewicz Krzysztof, Urbanek Petr, van der Meer Adriaan J, Van Vlierberghe Hans, Vella Stefano, Vince Adriana, Waheed Yasir, Waked Imam, Walsh Nicholas, Weis Nina, Wong Vincent W, Woodring Joseph, Yaghi Cesar, Yang Hwai-I, Yang Chung-Lin, Yesmembetov Kakharman, Yosry Ayman, Yuen Man-Fung, Yusuf Muhammed Aasim M, Zeuzem Stefan, Razavi Homie. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. The Lancet Gastroenterology & Hepatology. 2018;3(6):383–403. doi: 10.1016/S2468-1253(18)30056-6. - DOI - PubMed
-
- WHO. Hepatitis B Fact Sheet. Available at: http://www.who.int/mediacentre/factsheets/fs204/en/ (Accessed: May 2017) (2017).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
