

Next-generation characterization of the Cancer Cell Line Encyclopedia
- PMID: 31068700
- PMCID: PMC6697103
- DOI: 10.1038/s41586-019-1186-3
Next-generation characterization of the Cancer Cell Line Encyclopedia
Abstract
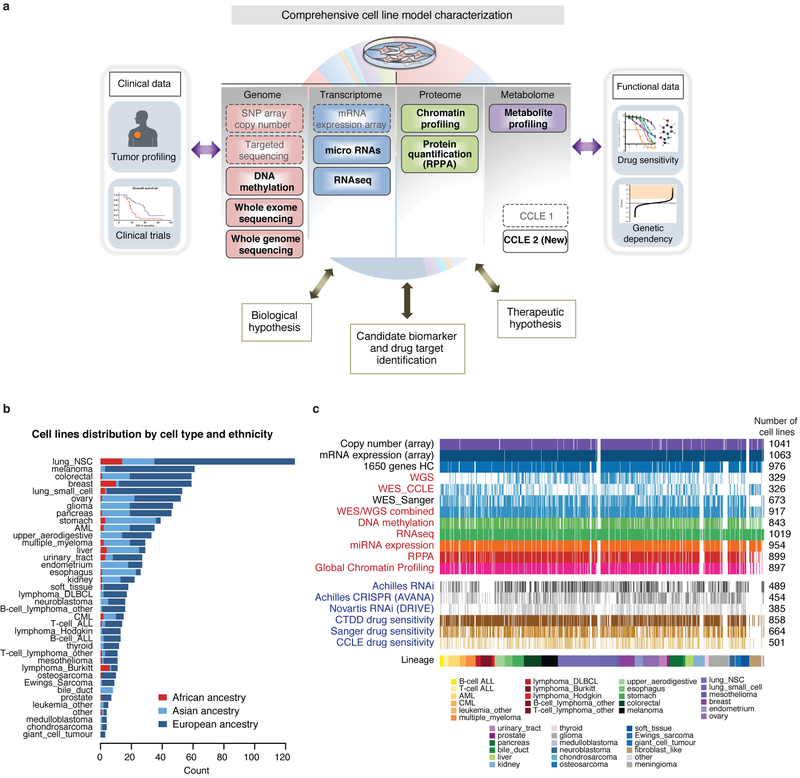
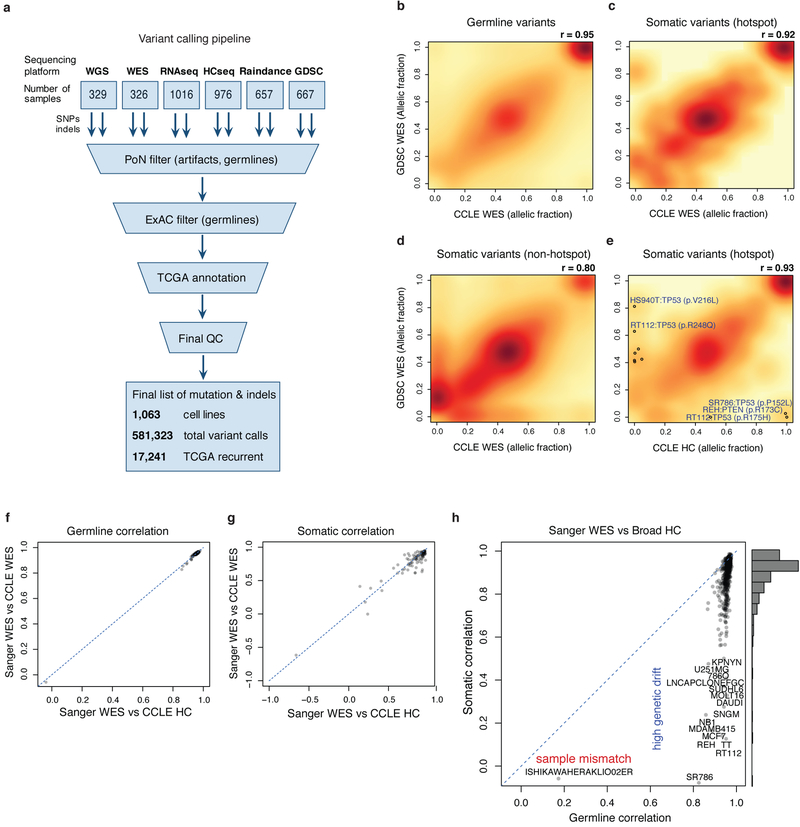
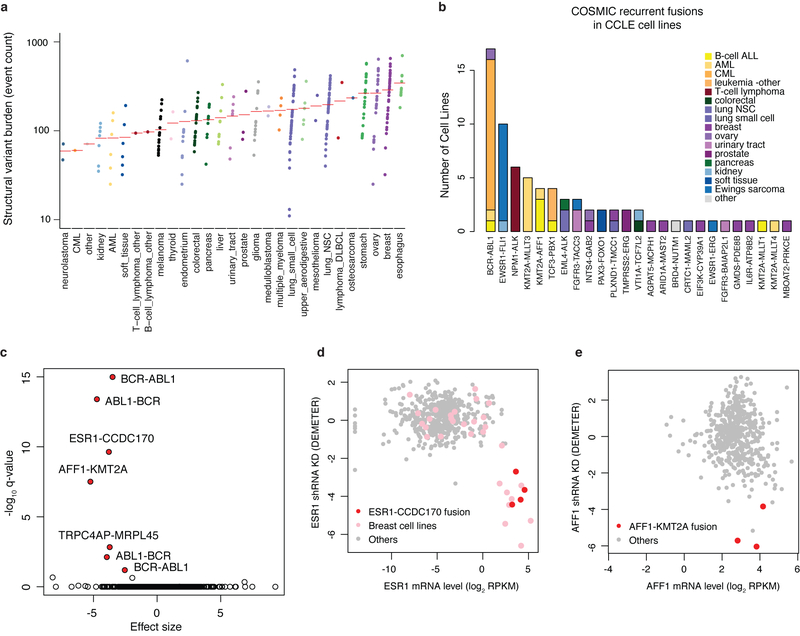
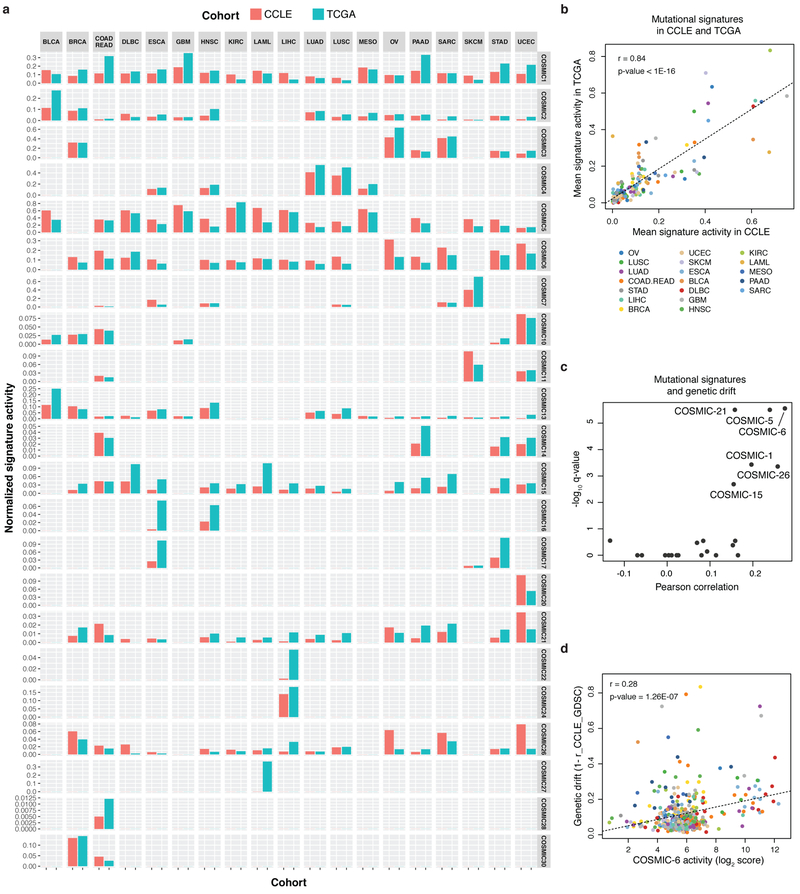
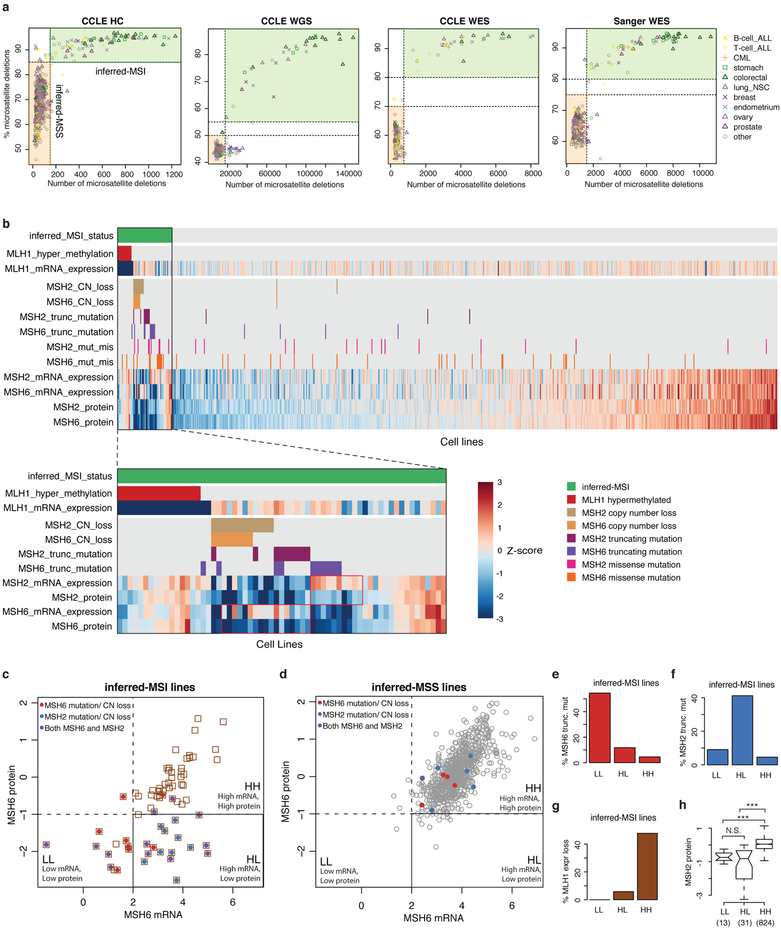
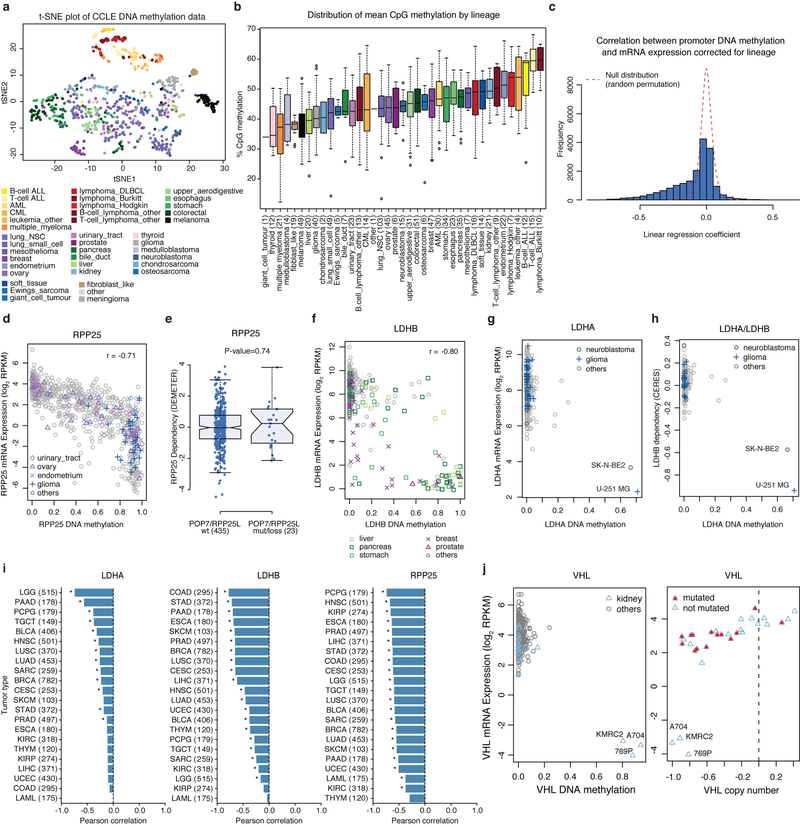
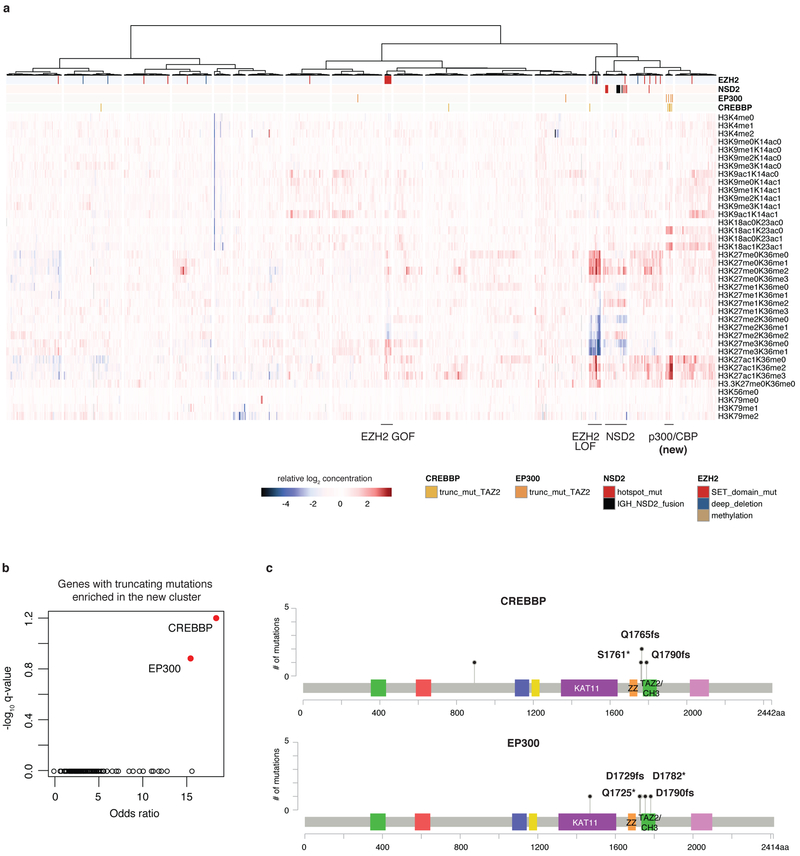
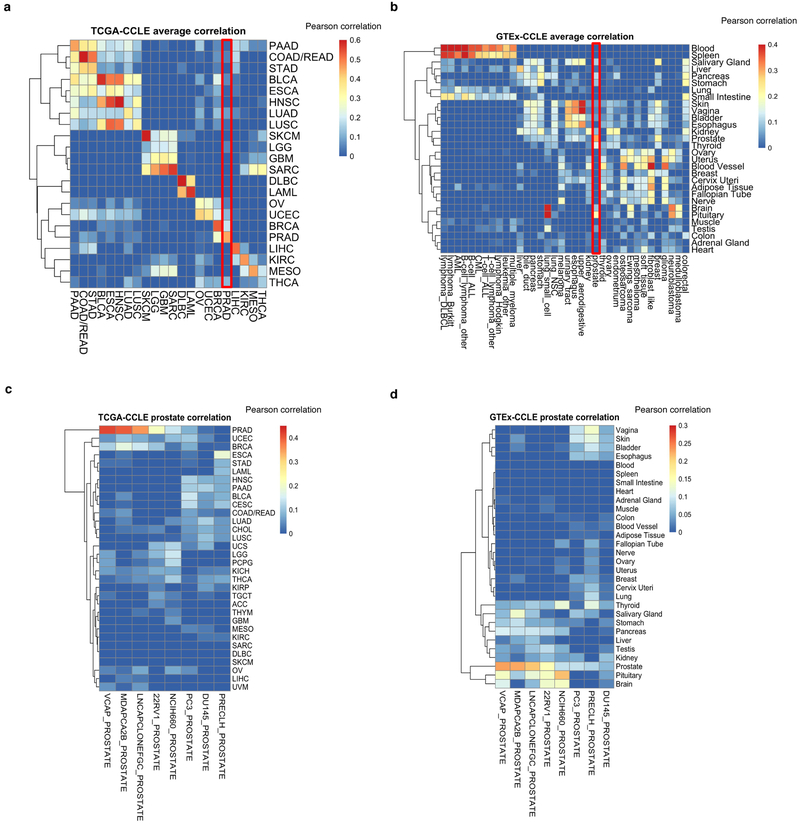
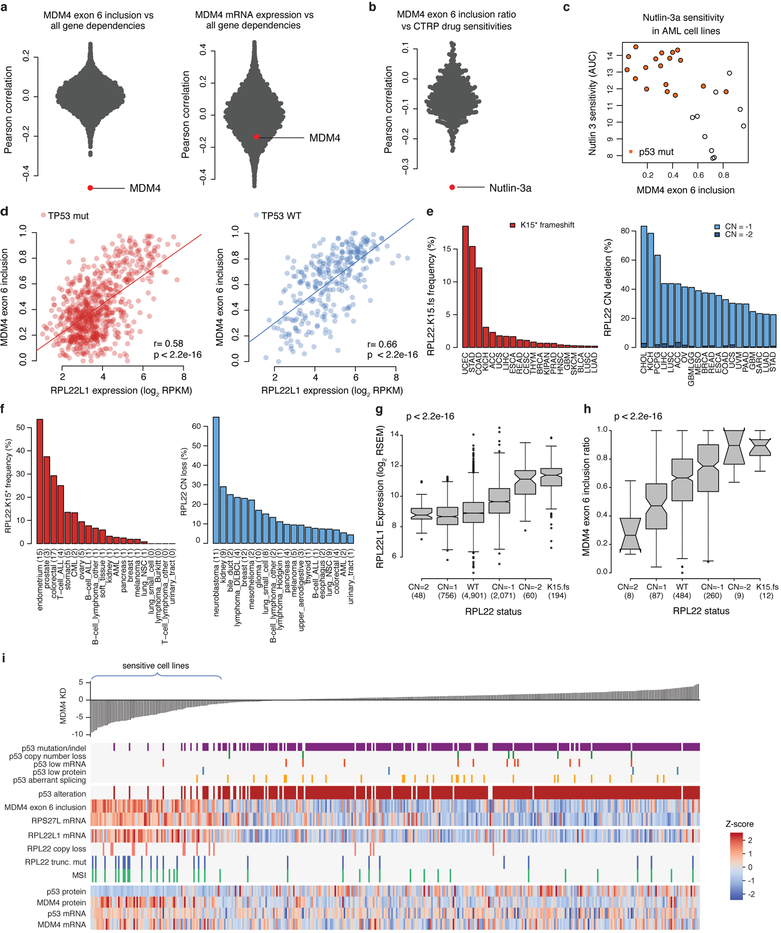
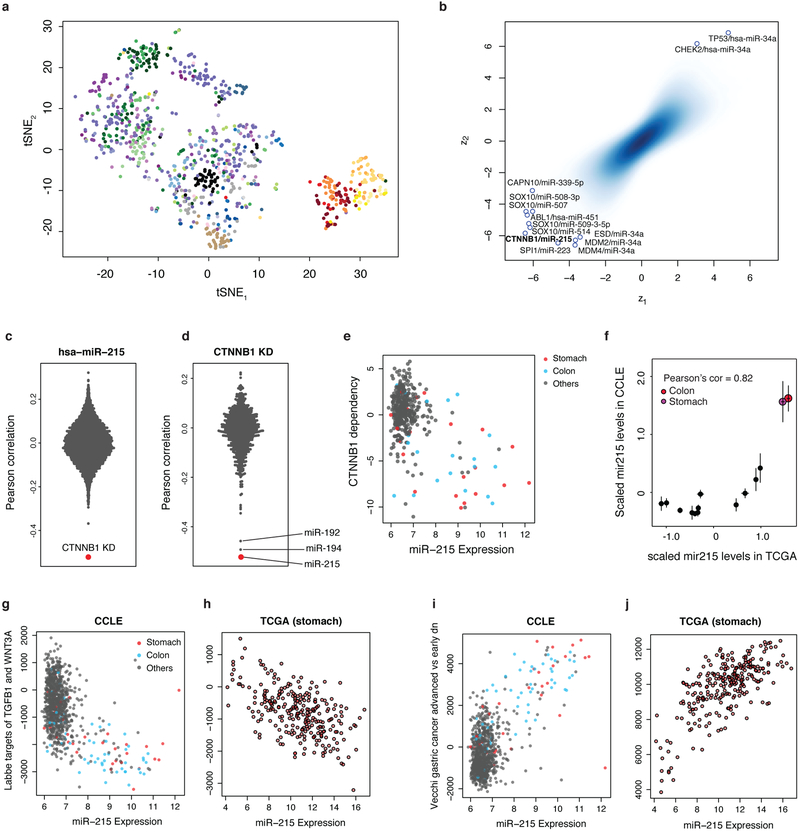
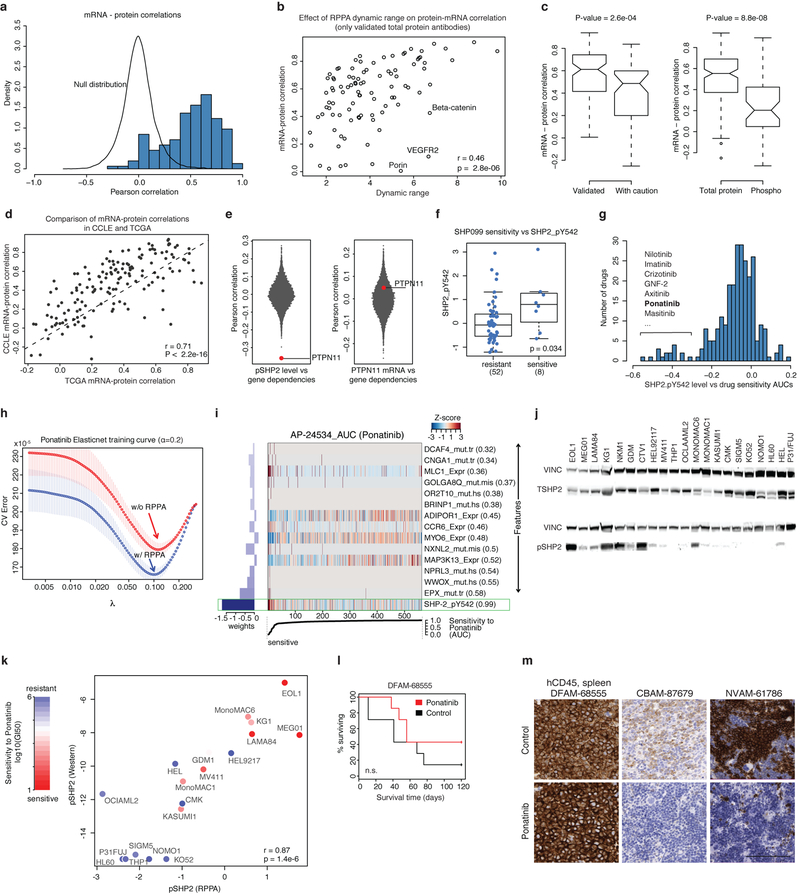
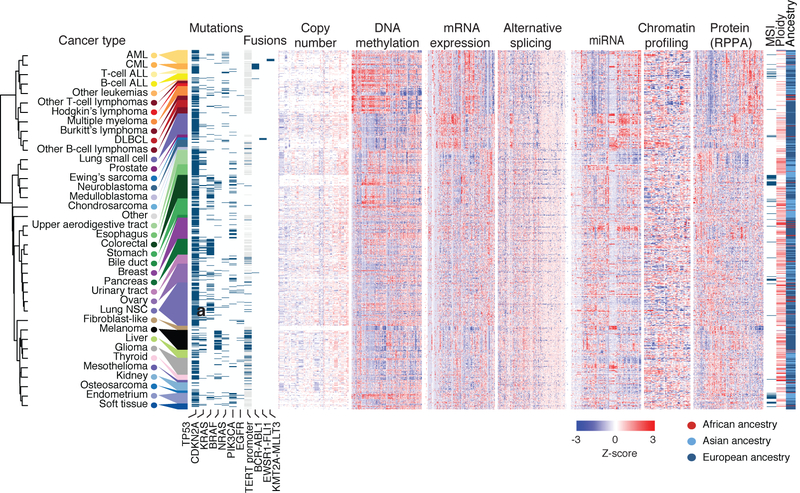
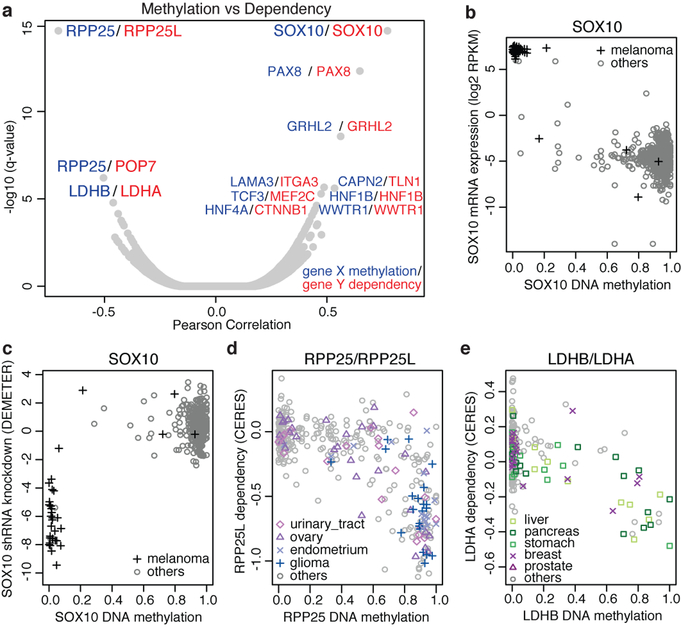
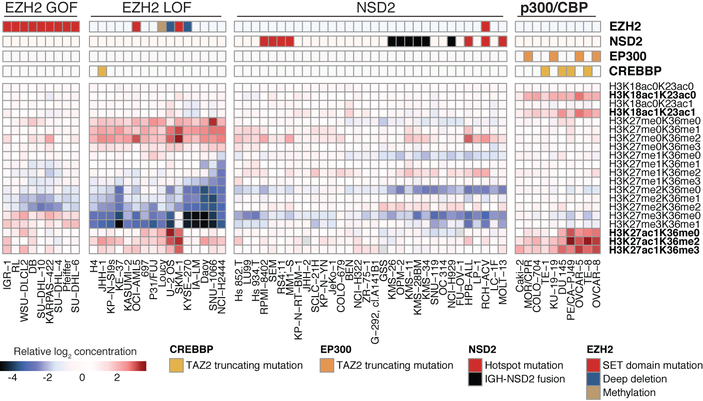
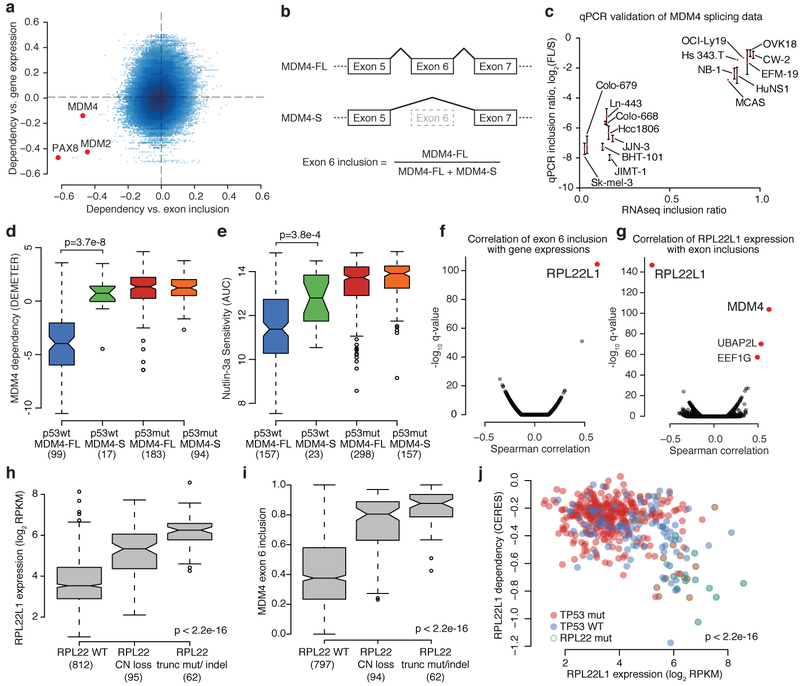
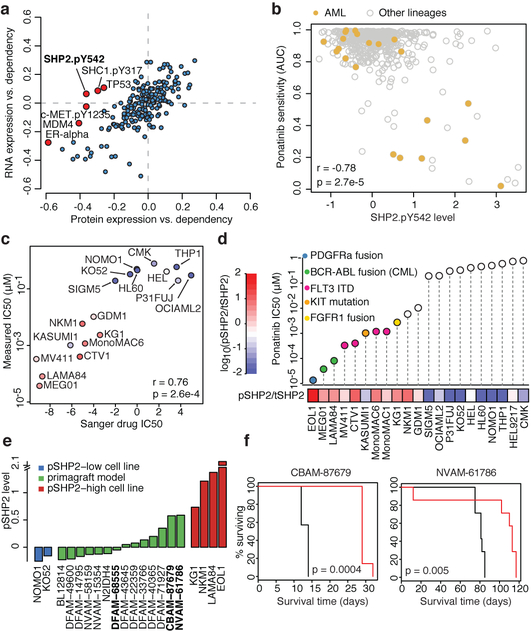
Large panels of comprehensively characterized human cancer models, including the Cancer Cell Line Encyclopedia (CCLE), have provided a rigorous framework with which to study genetic variants, candidate targets, and small-molecule and biological therapeutics and to identify new marker-driven cancer dependencies. To improve our understanding of the molecular features that contribute to cancer phenotypes, including drug responses, here we have expanded the characterizations of cancer cell lines to include genetic, RNA splicing, DNA methylation, histone H3 modification, microRNA expression and reverse-phase protein array data for 1,072 cell lines from individuals of various lineages and ethnicities. Integration of these data with functional characterizations such as drug-sensitivity, short hairpin RNA knockdown and CRISPR-Cas9 knockout data reveals potential targets for cancer drugs and associated biomarkers. Together, this dataset and an accompanying public data portal provide a resource for the acceleration of cancer research using model cancer cell lines.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- P30 CA016672/CA/NCI NIH HHS/United States
- U24 CA180922/CA/NCI NIH HHS/United States
- P50 CA217685/CA/NCI NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- R21 DA025720/DA/NIDA NIH HHS/United States
- U01 CA176058/CA/NCI NIH HHS/United States
- U54 CA224068/CA/NCI NIH HHS/United States
- R50 CA211461/CA/NCI NIH HHS/United States
- R01 CA219943/CA/NCI NIH HHS/United States
- R50 CA221675/CA/NCI NIH HHS/United States
- P50 CA098258/CA/NCI NIH HHS/United States
- U01 CA217842/CA/NCI NIH HHS/United States
- U24 CA210949/CA/NCI NIH HHS/United States
- R37 CA225191/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
